


AutoPepEx 介绍
自组装是大量分子在非共价相互作用的驱动下形成有序结构的过程,而短肽自组装材料因其优良的性质而广受关注。由于实验表征能力有限,分子动力学模拟成为研究短肽自组装的重要工具;为了探索广阔的短肽化学空间,机器学习和主动学习策略也被广泛应用。
AutoPepEx是一款结合PACE-ASM多尺度力场[1]分子动力学模拟和基于Uni-Mol分子表示学习方法[2]构建的主动学习策略来探索自组装短肽的工具,能对广阔的短肽化学空间进行快速探索,找到能发生自组装的短肽序列。
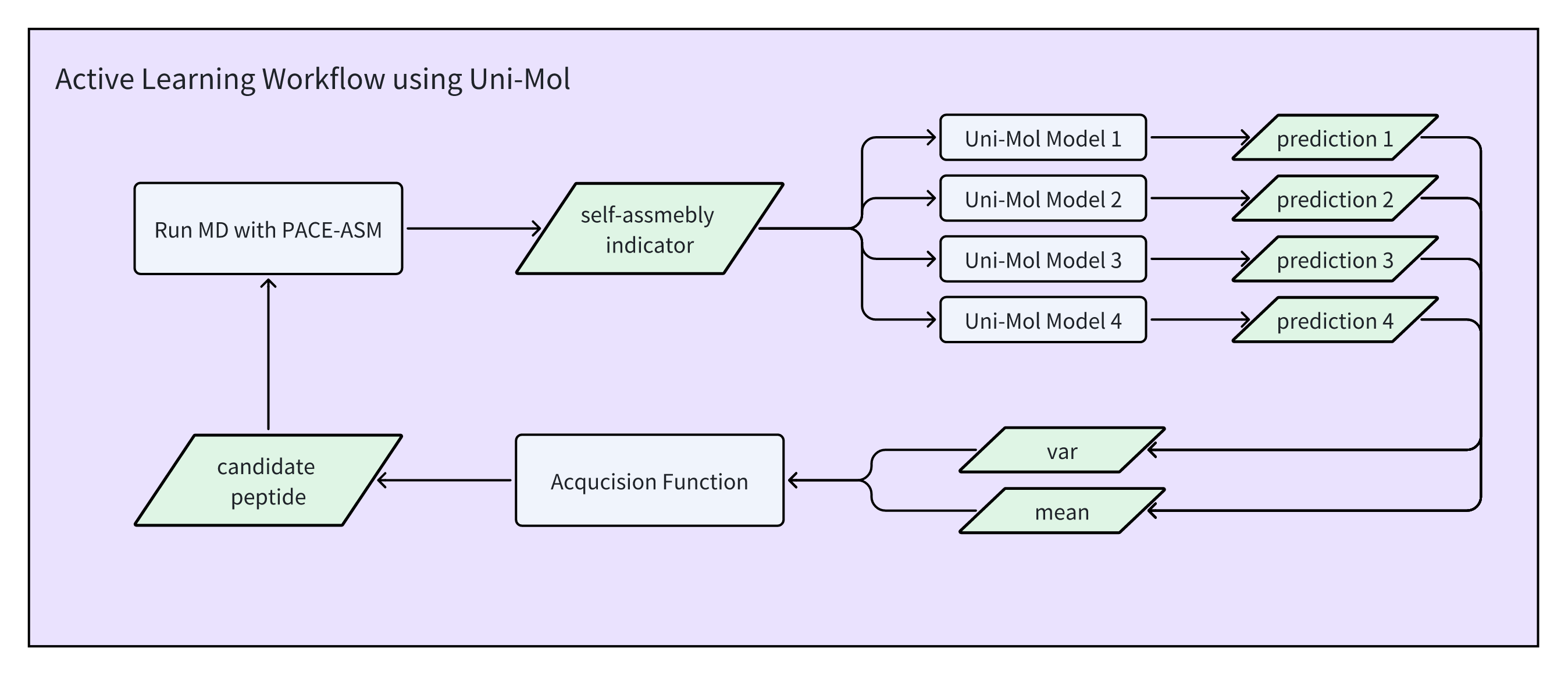
AutoPepEx的流程如上图所示,由以下步骤构成:
- (第一轮探索开始前)从短肽化学空间中随机选取一个短肽作为候选短肽。
- 计算模拟(labeling): 对候选短肽开展基于PACE-ASM多尺度力场的分子动力学模拟,基于模拟结果计算该短肽的自组装能力,并将该短肽及其自组装能力加入到训练数据集中;
- 模型训练(training): 以训练集中的短肽及其自组装能力训练4个独立的Uni-Mol模型;
- 主动探索(exploring): 使用训练好的4个模型对未探索短肽的自组装能力进行预测,并获取这4个模型对同一个短肽预测值的均值和方差,随后使用期望提升(EI)算法挑选下一个候选短肽;
- 返回计算模拟(labeling)步骤,重复以上操作,直至计算模拟次数用完,循环结束。使用训练得到的模型对整个短肽化学空间进行预测,给出全空间的预测结果。
v1.1版本
在v1.1版本中,我们增加了基于Uni-Mol预测短肽自组装能力的功能。您可以在提交任务的首页选择"prediction|Predict self-assembly ability of peptides using Uni-Mol"来使用这个功能。
由于通过模型预测会比开展分子动力学模拟快得多,所以我们建议您在使用AutoPepEx的时候,可以先通过Uni-Mol模型预测您感兴趣的短肽序列的自组装能力,然后再挑选模型预测比较好的那些序列进行长时间的PACE-ASM分子动力学模拟,来获取准确的自组装能力评估。
v1版本
由于开展整个主动学习流程非常消耗算力。因此在v1版本中,我们仅提供了单个短肽的计算模拟方法。您只需输入您感兴趣的短肽序列,设定短肽聚集体的数目和合适的模拟时长,就能开展基于PACE-ASM多尺度力场的分子动力学模拟。得到模拟结果后,您能够观察到短肽自组装的动力学过程和短肽自组装体的微观形貌,并能获取短肽的自组装能力指标和聚集能力指标AP。
其中,PACE-ASM多尺度力场是一种专门面向短肽自组装体系模拟进行优化的多尺度力场。在PACE-ASM多尺度力场下,蛋白模型则采用联合原子模型表示,仅简化了部分氢原子,保留了绝大多数蛋白原子细节;溶剂被简化为粗粒化力场MARTINI溶剂粒子,一个溶剂粒子代表四个水分子。这样的建模方式使PACE-ASM多尺度力场相比全原子力场有着更高的效率,可以增大模拟的时空尺度;相比粗粒化力场有原子细节,可以反映短肽自组装过程中的分子间相互作用。
参考文献
[1] Cai, Xiang, and Wei Han. Journal of Chemical Information and Modeling 62.11 (2022): 2744-2760. [2] Zhou, Gengmo, et al. ICLR 2023 (2023).