


致谢:
原notebook链接:https://bohrium.dp.tech/notebooks/45986312168
Na的基本介绍
1. 发现历程
钠作为一种广泛存在的元素,其发现历史却主要于电池的发展有关,在19世纪初,伏特(Volta A.G. 1745—1827,意)发明了电池后,各国化学家纷纷利用电池分解水成功。英国化学家戴维(Davy H. 1778—1829)坚持不懈地从事于利用电池分解各种物质的实验研究。他希望利用电池将苛性钾分解为氧气和一种未知的“基”,因为当时化学家们认为苛性碱也是氧化物。它先用苛性钾的饱和溶液实验,所得的结果却和电解水一样,只得到氢气和氧气。后来他改变实验方法,电解熔融的苛性钾,在阴极上出现了具有金属光泽的、类似水银的小珠,一些小珠立即燃烧并发生爆炸,形成光亮的火焰,另一些小珠不燃烧,只是表面变暗,覆盖着一层白膜。他把这种小小的金属颗粒投入水中,即起火焰,在水面急速奔跃,发出刺刺的声音。就这样,戴维在1807年10月6日发现了金属钾,几天之后,他又从电解苛性钠中获得了金属钠。
戴维将钾和钠分别命名为Potassium和Sodium,因为钾是从草木灰(Potash),钠是从天然碱——苏打(Soda)中得到的,它们至今保留在英文中。钾和钠的化学符号K,Na分别来自它们的拉丁文名称Kalium和Natrium。
2. 基本性质 钠是一种质软,银白色,有金属光泽的金属,具有良好的导电、导热性,密度比水小(0.968g/cm3),比煤油大,熔点较低(97.72℃),沸点883℃。特征光为黄色(能盖住很多特征光)。
相对原子量22.98976928,原子半径180 pm,范德华半径250 pm,鲍林电负性0.93,单质晶格为体心立方晶格,晶格常数:a: 429.06 pm b: 429.06 pm c: 429.06 pm α: 90.000° β: 90.000° γ: 90.000°。
DFT原理
1.密度泛函起源
Thomas-Fermi模型,即将电子密度泛函n(r)作为变量,来表示体系系统的总能量,即:
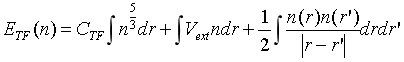
并有约束条件:

从而可以求解总能量泛函的极小值,即可得到系统的基态电子密度,如采用拉格朗日乘子法:
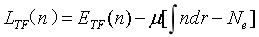
对拉格朗日函数变分求零并自洽迭代计算,最终可以得到系统基态电子密度和基态能量。
2.Hohenberg-Kohn理论
Hohenberg-Kohn定理1:对于处在任一外势场Vext(r)下的有相互作用粒子组成的系统,外势场Vext(r)由基态粒子密度n0(r)唯一确定,其中Vext(r)可以相差一个常数
Hohenberg-Kohn定理2:对于任意给定的外势场Vext(r),有能量泛函E(n)当电子密度n(r)取到系统基态电子密度n0时,该能量泛函E(n)取到系统基态能量
3.Kohn-Sham方程
Kohn-Sham假设:对于一个多体系统,总是可以找到一个对应的无相互作用的单体系统,这两个系统有同样的基态电子密度。从而在单体系统中电子可以用单电子波函数描述,而这两个系统之间的能量差来源于实际多体系统中电子之间的相互作用并归于“交换关联泛函”。
对于一个Ne个电子的系统,KS系统电子密度为:

并由Kohn-Sham方程自洽迭代得到系统能量:

4.交换关联泛函
交换关联泛函Exc描述了单粒子近似下系统与真实系统之间的能量差,因此Exc的精度很大程度上决定了DFT计算的精度,一般将交换关联泛函定义为:

这里介绍最常用的PBE泛函:
定义无量纲的密度梯度:

定义自旋极化增强因子:

从而可以写出PBE交换能的形式:
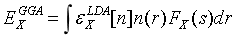
定义自旋标度因子:
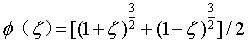
并定义无量纲的密度梯度:
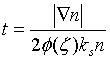
PBE关联能

ABACUS介绍
ABACUS(原子算筹)作为一款国产的电子结构软件,可用于对材料进行密度泛函理论(Density Functional Theory,简称 DFT)的第一性原理(first-principles)计算,其主要核心功能之一是电子自洽迭代计算(Self Consistent Field,简称SCF),在给定材料微观层面的晶胞和原子位置的条件下,我们可以通过 SCF 流程的计算获得电子结构的总能量、能级、电子波函数、电子密度等关键信息,这些信息可用于进一步计算得出材料体系的其它性质。
ABACUS软件使用——收敛性测试
准备文件
自洽迭代计算需要准备三个主要文件即
INPUT
KPT
STRU
三个最基本的文件作为 ABACUS 的基本输入文件。STRU 文件提供元素、晶格、原子的基本结构信息;KPT 文件提供周期性边界条件下布里渊区采样的网格设置。INPUT 文件主要提供计算所需的各种设置参数,在图1里我们分成了五部分且将在下面详细介绍。
以及需要准备轨道和赝势文件。相对应的轨道和赝势文件可以在https://github.com/kirk0830/ABACUS-Pseudopot-Nao-Square/tree/main/download下载,并和INPUT等文件放在一起
bohr/NaDFT-2pm8/v1/Na/SCF ├── INPUT ├── KPT ├── Na_ONCV_PBE-1.0.upf ├── Na_gga_8au_100Ry_4s2p1d.orb ├── OUT.Na │ ├── INPUT │ ├── STRU_READIN_ADJUST.cif │ ├── STRU_SIMPLE.cif │ ├── istate.info │ ├── kpoints │ ├── running_scf.log │ └── warning.log ├── STRU └── time.json 1 directory, 13 files
INPUT文件:
INPUT_PARAMETERS #Parameters (1.General) suffix Na calculation scf symmetry 1 pseudo_dir . orbital_dir . basis_type pw ecutwfc 100 #Parameters (2. SCF iterations) scf_nmax 100 scf_thr 1e-8 #Parameters (3. Solve KS equation) nbands 11 ks_solver cg #Parameters (4.Smearing) smearing_method gauss smearing_sigma 0.01 #Parameters (5.Mixing) mixing_type broyden mixing_beta 0.7 mixing_gg0 0S
suffix:suffix 是用户可以自定义的后缀,运行 ABACUS 可执行程序之后,输入文件所在的文件夹里会生成一个包含大部分运行信息的 OUT.suffix 文件夹。
calculation:设置本次计算类型,例如本次计算 "scf"(自洽电子结构计算)。scf((Self Consistent Field)、relax、cell-relax、md(Molecular Dynamics)是较常用的四类计算。
symmetry:是否考虑对称性,有如下三个选项
-1 不进行对称性分析 0 仅考虑时间反演对称性 1 进行对称性分析
如果打开对称性(设置为 1),布里渊区 k 点可以根据对称性进行简化处理,若体系有对称性,则可以减少所需计算的 k 点。因为每个 k 点都会进行一次 Kohn-Sham 方程的求解,对称性分析后若 k 点减少则将提升计算效率。本次计算中开启对称性分析,设置 "1",关于对称性分析的测试还在进一步完善,如果计算结果奇怪,建议设置成 "0"之后再进行计算,比较结果是否一致。
pseudo_dir:计算中需要使用赝势来近似离子和电子相互作用势能,为计算提供赝势文件。pseudo_dir指定 STRU 文件中赝势文件所在的目录。本次计算将赝势和轨道文件都与 INPUT、STRU、KPT 文件放在一起,因此填入 ".",表示处在当前文件夹中。
orbital_dir:本次计算将采取 pw(平面波轨道)基组(后文 basis_type 中设置)。若采用 LCAO 基组进行计算,则需要提供作为基组的轨道文件,在 STRU 文件中指定轨道文件所在的目录。若选用 pw 基组,则不需要轨道文件,不需要填写此参数。
basis_type:计算的基组(指描述电子波函数的基函数),在 ABACUS 中常用的有两种:
pw:平面波基组(由于基矢量更完备,pw 计算将更准确,同时计算时间也更长)
LCAO:局域原子轨道基组(没有 pw 准确,但效率高)
ecutwfc:平面波函数的能量截止(单位:Ry),在平面波基组里是很重要的一个参数,其大小决定着作为基矢的平面波函数的个数多少,而基矢的多少,也决定了计算精度的高低。
scf_nmax:针对每个离子构型,scf 电子迭代的最大次数为 scf_nmax,可设为"50"或"100" 次。半导体和绝缘体是较容易收敛的体系,一般 20 步 scf 以内即可达到收敛,对于金属体系或者费米面较为复杂的体系,有可能 50 步仍未收敛。注意,如果做 relax, cell-relax, 或者 md 的时候发现有某些 scf 迭代次数达到最大时仍未达到收敛条件,建议先把计算停下来弄清楚原因后再继续算,有可能是因为离子构型或者 k 点等参数的设置不合理引起的。
scf_thr:代表两个相邻电子迭代步之间的电荷密度误差,也是 SCF 计算中判断是否收敛、完成计算的标准,对于 LCAO,一般设置为 "1e-7",可认为精度足够。对于 pw,建议设置 1e-8 或 1e-9 的精度。
nbands:计算的 Kohn-Sham 轨道数目,在本次计算中无磁性,参数 nspin 取 1(默认值),程序目前采取 0.5max(1.2occupied_bands, occupied_bands + 10) 计算 nbands。
ks_solver:在不同基组中展开哈密顿矩阵的对角化方法,对于 pw,可以选择 cg(Conjugate Gradient,默认方法),bpcg(还不是太稳定、测试中),dav(Davidson 算法);对于 LCAO,可以选择 genelpa(默认值),scalapack_gvx(Scalable Linear Algebra PACKage)。如果选用 LCAO 基组,ks_solver 可设置为"genelpa"。
smearing_method:对于金属体系或者费米面附近较复杂的体系,电子自洽迭代方法往往不容易收敛,这个时候可以选择给电子提供一个光滑的占据函数,用来调节程序计算电荷密度和总电子数时候的电子占据函数,这对于费米面附近的电子态尤为重要。我们这里称为 "smearing method",或者称为展宽方法。具体来说,对于金属体系可以选择 mp(methfessel-paxton),mp2(2-nd methfessel-paxton) ,也可以使用 gauss(也可以写作 gaussian)。因为我们计算金刚石 Si 具有半导体性质,所以 smearing 方法基本不起作用,默认可以选择 gauss。此外,非导体计算可以选择 fixed,半导体和非导体也可以选择 fd(Fermi-Dirac)方法。
smearing_sigma:给定展宽方法的能量范围(单位:Ry),默认是 0.015 Ry,我们按照通常情况给定 "0.01"
KPT文件
K_POINTS 0 Gamma 4 4 4 0 0 0
STRU文件
ATOMIC_SPECIES Na 22.9898 Na_ONCV_PBE-1.0.upf NUMERICAL_ORBITAL Na_gga_8au_100Ry_4s2p1d.orb LATTICE_CONSTANT 1.8897261258369282 LATTICE_VECTORS 4.2905998230 0.0000000000 0.0000000000 0.0000000000 4.2905998230 0.0000000000 0.0000000000 0.0000000000 4.2905998230 ATOMIC_POSITIONS Direct Na 0.0000000000 2 0.0000000000 0.0000000000 0.0000000000 1 1 1 mag 0.0 0.5000000000 0.5000000000 0.5000000000 1 1 1 mag 0.0
STRU文件中对应计算采用的原子名称、分子量、以及轨道和赝势文件的名称
另外包含原子单质晶格的相关数据,该数据可以通过vasp的.vasp文件进行转换
自洽迭代计算
准备好上述INPUT KPT STRU文件后将三者和轨道文件以及赝势文件放在同一个目录下输入:
mpirun -n 2 abacus
其中 -n 2表示调动2个核计算,一个较为粗糙、但基本不会造成计算资源浪费的选取 n 值原则是:体系有多少个原子,不要用超过这个原子个数太多的总核数进行并行计算。例如,体系如果有 16 个原子,不要用远大于 16 的总核数进行计算,一般取 16 的总核数或者更少就够了,具体需要测试。
输出结果后,计算结果存储在根目录下.OUT文件中,输入:
grep ETOT OUT.Na/running_SCF.log
可以得到系统总能量
!FINAL_ETOT_IS -2318.365132995114 eV
Ecut和K点收敛性测试
计算ecut和K点的收敛性,我们需要不断更改INPUT或者KPT文件中的ecut值或者K点数目**“n n n **
更改后按照上述步骤计算系统总能量并给出能量变化趋势图(比如导出数据后利用orgin画图),这里我们ecut变化范围为20-100步长为10,K点取值2,4,6,8

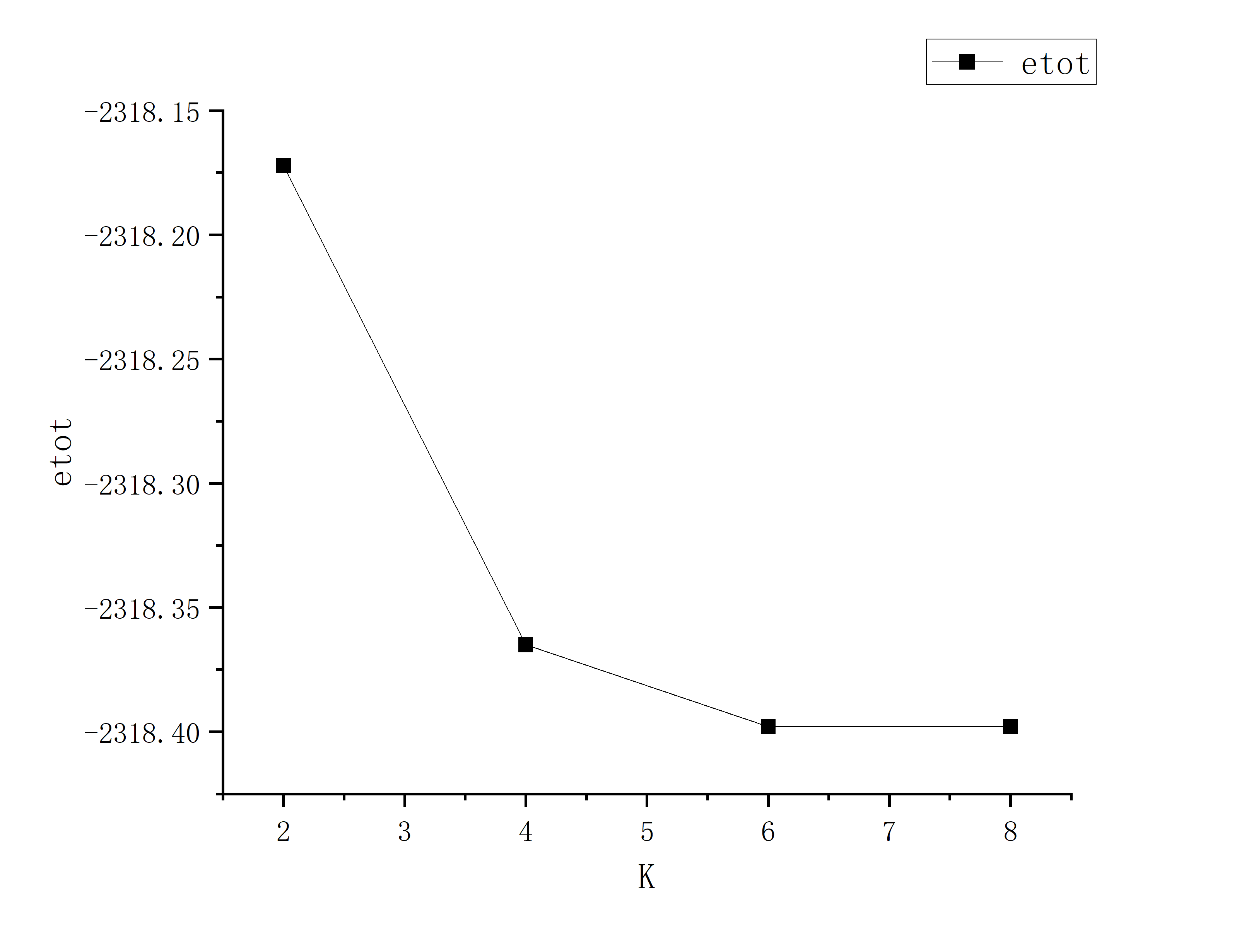
可以看到K数在6之后就可认为达到收敛以及可以看到ecut在40-60中间就可以认为达到收敛
ABACUS软件使用——结构弛豫
Na单质结构弛豫
对Na的晶格进行结构弛豫,首先需要更改INPUT中的计算模式,将calcuation更改为cell_relax,并准备KPT和STRU文件 如:
INPUT_PARAMETERS suffix Na ntype 1 nelec 0.0 pseudo_dir ./ orbital_dir ./ ecutwfc 100 # Rydberg scf_thr 1e-4 # Rydberg basis_type pw calculation cell-relax # this is the key parameter telling abacus to do a optimization calculation force_thr_ev 0.01 # the threshold of the force convergence, in unit of eV/Angstrom stress_thr 2 # the threshold of the stress convergence, in unit of kBar relax_nmax 100 # the maximal number of ionic iteration steps out_stru 1 out_mul 1 nspin 2
输入:
mpirun -n 2 abacus
最终弛豫后的结果输出在**.OUT** 文件中 STRU_NOW和STRU_READIN_ADJUST.cif中
data_none _audit_creation_method generated by ABACUS _cell_length_a 3.69437 _cell_length_b 3.69437 _cell_length_c 3.69437 _cell_angle_alpha 109.471 _cell_angle_beta 109.471 _cell_angle_gamma 109.471 loop_ _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z Na 0 0 0
data_none _audit_creation_method generated by ABACUS _cell_length_a 3.7158 _cell_length_b 3.7158 _cell_length_c 3.7158 _cell_angle_alpha 109.471 _cell_angle_beta 109.471 _cell_angle_gamma 109.471 loop_ _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z Na 0 0 0
(relax中计算的是Na的原胞),可以查询资料:,Na单质为体心立方结构,其原胞三个边为体对角线的一半,因此通过几何关系可知三个基矢夹角为109.5°,由于Na的晶胞常数为429pm,计算可得其体对角线一半为371pm(3.71,计算出结果为3.694)计算结果近似但是偏小
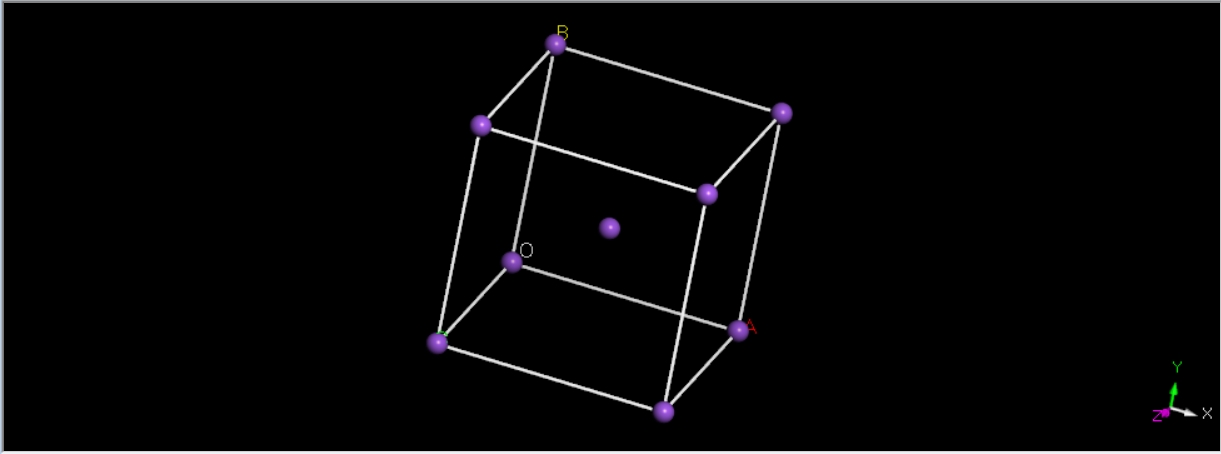
Na2O结构优化
准备INPUT KPT STRU文件,需要注意的是由于Na2O有两种原子因此STRU和INPUT文件和单质略有不同
INPUT:
1.INPUT_PARAMETERS
2.suffix Na2O
3.ntype 2
4.nelec 0.0
5.pseudo_dir ./
6.orbital_dir ./
7.ecutwfc 100 # Rydberg
8.scf_thr 1e-4 # Rydberg
9.basis_type pw
10.calculation cell-relax # this is the key parameter telling abacus to do a optimization calculation
11.force_thr_ev 0.01 # the threshold of the force convergence, in unit of eV/Angstrom
12.stress_thr 2 # the threshold of the stress convergence, in unit of kBar
13.relax_nmax 100 # the maximal number of ionic iteration steps
14.out_stru 1
15.out_mul 1
16.nspin 1
17.#Parameters (5.Mixing)
18.mixing_type broyden
19.mixing_beta 0.8
20.mixing_gg0 0S
STRU:
1.ATOMIC_SPECIES
2.Na 22.9898 Na_ONCV_PBE-1.0.upf
3.O 15.9994 O_ONCV_PBE-1.0.upf
4.
5.NUMERICAL_ORBITAL
6.Na_gga_10au_100Ry_2s1p.orb
7.O_gga_10au_100Ry_2s2p1d.orb
8.
9.LATTICE_CONSTANT
10.1.8897261258369282
11.
12.LATTICE_VECTORS
13.3.9244000912 0.0000000000 0.0000000000
14.1.9622000456 3.3986301736 0.0000000000
15.1.9622000456 1.1328767245 3.2042592566
16.
17.ATOMIC_POSITIONS
18.Direct
19.Na
20.0.0000000000
21.2
22.0.25000000000 0.2500000000 0.2500000000 1 1 1 mag 0.0
23.0.75000000000 0.7500000000 0.7500000000 1 1 1 mag 0.0
24.O
25.0.0000000000
26.1
27.0.00000000000 0.0000000000 0.0000000000 1 1 1 mag 0.0
KPT:
1.K_POINTS
2.0
3.Gamma
4.7 7 7 0 0 0
输入:
mpirun -n 2 abacus
即可开始计算,最终弛豫后的结果输出在**.OUT** 文件中 STRU_NOW
data_none _audit_creation_method generated by ABACUS _cell_length_a 3.96684 _cell_length_b 3.96684 _cell_length_c 3.96684 _cell_angle_alpha 60 _cell_angle_beta 60 _cell_angle_gamma 60 loop_ _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z Na 0.25 0.25 0.25 Na 0.75 0.75 0.75 O 3.06118e-41 3.06118e-41 3.8877e-39
氧化纳为反萤石结构即fcc堆积,原胞三个角均为60°,原胞晶格长度为晶胞面对角线,查阅可知,氧化纳晶胞边长为550pm故原胞实际为389pm(3.89计算结果为3.97)计算结果接近但偏大