锆(Zr)的发现及背景: 锆的名字来源于矿物锆石,锆石在几个世纪前就被用作珠宝,并在《圣经》中被提及。德国化学家马丁·海因里希·克拉普罗特于1789年首次识别出锆元素,而瑞典化学家贝齐利阿斯于1824年首次提纯出纯锆。
物理化学性质: 锆是银灰色、有金属光泽、延展性强的金属,密度为6.49g/cm³,熔点为2125K,沸点为4650K。它具有很强的抗腐蚀能力,在低纯度下则较脆。锆的电子结构具有六方紧密堆积的晶体结构,在863°C时转变为体心立方晶体结构。
同位素: 锆有五种天然同位素(90Zr、91Zr、92Zr、94Zr、96Zr),其中90Zr最常见,占51.45%。此外,还有28种人工合成的锆同位素。
常见化合物: 锆的常见化合物包括二氧化锆(ZrO2)、碳化锆(ZrC)、氮化锆(ZrN)以及各种卤化物(如ZrF4、ZrCl4)。这些化合物在陶瓷、耐火材料、催化剂等领域有广泛应用。
主要用途: 宝石:由于锆石的美丽外观,常被用作装饰品。 陶瓷和耐火材料:硅酸锆和二氧化锆广泛用于建筑陶瓷、耐火材料等。 核工业:核级锆用于核反应堆的结构材料。 化工设备:工业级锆因其优异的抗腐蚀性能,用于化工设备和电子行业。 其他用途:锆还用于火器燃烧剂、合金添加剂、冶金脱氧剂等。 总结 锆是一种具有多种优良性质的金属,其在宝石、陶瓷、核工业、化工等领域都有重要应用。通过复杂的提纯和加工工艺,锆被制成各种产品,满足不同行业的需求。
DFT算法基本原理介绍
密度泛函理论(density functional theory,简称DFT)是一种研究多电子体系电子结构的量子力学方法,在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。
密度泛函理论的主要目标就是用电子密度取代波函数作为研究的基本量。
理论依据:Hohenberg-Kohn定理
1、对任意相互作用的电子系统,处在外部势场V中,则该外部势场V可由基态电子密度n(r)唯一决定(除了相差一个常数)。
2、对任一给定的外势Vext(r),可定义关于电子密度的普适性泛函,其中,基态(非简并)电子密度使得此能量泛函取极小值。
这两个定理理论上可以精确地描述多电子系统,其给出了对应关系的存在,但没有给出对应关系的具体形式。
Kohn-Sham方程
密度泛函理论最普遍的应用是通过Kohn-Sham方程实现的。Walter Kohn提出Hohenberg-Kohn定理的后一年,与沈吕九(Lu Jeu Sham)提出了著名的Kohn-Sham假设:
有相互作用系统的基态电荷密度,可以被一个无相互作用系统的基态电荷密度表示出来。同时推导出了Kohn-Sham方程:
其中是Kohn-Sham哈密顿量,是Kohn-Sham波函数,
是外势,包括原子核引力和外加电场,
是哈特里势,描述了电子间的库仑排斥作用,
(r) 是交换关联势能,包括交换能和关联能,
是Kohn-Sham波函数的能量本征值。
可以通过Kohn-Sham波函数构造电子密度:
交换关联泛函近似
此外,交换关联泛函的近似也十分重要,其好坏决定了DFT方法的精度和效率。 常见的近似有Kohn-Sham给出的局域密度近似(Local Density Approximation,简称LDA),假设实空间每一点r的交换关联泛函用均匀自由电子气的交换关联泛函来代替,即是局域的。还有广义梯度近似(GGA),其引入了电子密度梯度的修正。以及杂化泛函(Hybrid Functionals)等。
KS方程求解——电子自洽迭代
科恩-沈吕九方程的求解需要用自洽方法。通常首先假设一个初始的 𝑛(𝑟), 然后计算对应的𝑉𝑠 并求解科恩-沈吕九方程中的𝜙𝑖。进而可以计算出新的密度分布,并开始新一轮计算。此过程不断重复,直到计算结果收敛。
DFT算法可以用于计算分子结构(确定基态结构,预测几何结构),电子结构(电子密度分布,能带结构,态密度),分子和材料的光学性质,磁性,分子动力学以及反应机理等。
ABACUS软件使用方法
ABACUS(原子算筹)是国产开源密度泛函理论软件,支持基于量子力学的密度泛函理论计算(DFT),也可结合经验势或者DPMD进行模拟。
目前支持的主要功能有采用平面波或数值原子轨道作为基矢量,支持多种并行方案,结合模守恒赝势进行Kohn-Sham方程迭代求解及各种形式的DFT运算和AI辅助算法等。
Zr元素的收敛性测试:
平面波收敛性测试 :
使用ABACUS软件常见地需要准备INPUT,STRU,KPT三种文件,此外,在计算过程中可能会用到赝势文件,可以在https://github.com/kirk0830/ABACUS-Pseudopot-Nao-Square/tree/main/download/pseudopotentials
此文档中找到所需元素的赝势,如果使用lcao基组还需要原子轨道文件。赝势文件天然包含交换关联泛函,目前最常见的是PBE交换关联泛函。
按照此教程的方法https://mcresearch.github.io/abacus-user-guide/abacus-pw.html配置INPUT,STRU,KPT文件。
INPUT 文件中设置 ecut 分别为 60-120,间隔为 10,其余不变。 STRU 文件中按照cell-relax 得出的结果设置结构。 KPT 文件中 K 点个数设置为 444。随后可利用脚本提取数次结果最后的基态能量,最后得到的结果如图所示。发现在ecut=90时开始收敛。
k-point的收敛性测试:
随后将ecut值设为100,设置k的值分别为2-8,间隔为1,其余不变,再利用脚本提取得到的基态能量。最终得到的结果如图所示。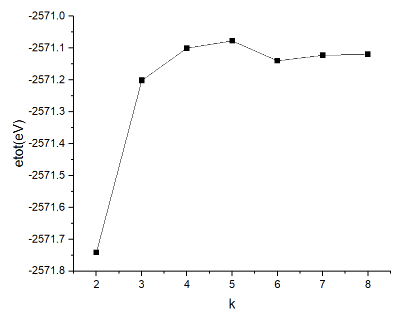
可以看出从k=6开始,etot的值开始收敛。
自洽迭代运算
进入INPUT、KPT、STRU三个文件所在的目录,执行以下命令即可开始计算。运行成功后,输出文件将在同目录下OUT.N文件夹中,其中的running_scf.log文件可以查找到晶体的总能量。
# 进入工作文件夹
cd ./ABACUS/U_PW/SCF
# OMP_NUM_THREADS=1 表示使用单线程,如果你的机器配置比较高,可以使用多线程,比如 4 线程,就可以写成 OMP_NUM_THREADS=4
# mpirun -n 后面的数字表示计算所使用的 CPU 核心数,这里使用 2 个核心,你可以根据你的机器配置进行修改。
OMP_NUM_THREADS=1 mpirun -n 2 abacus
附录:
INPUT文件
INPUT_PARAMETERS
#Parameters (1.General)
suffix Zr
calculation scf
symmetry 1
pseudo_dir .
orbital_dir .
basis_type pw
ecutwfc 100
#Parameters (2. SCF iterations)
scf_nmax 100
scf_thr 1e-8
#Parameters (3. Solve KS equation)
nbands 22
ks_solver cg
#Parameters (4.Smearing)
smearing_method gauss
smearing_sigma 0.01
#Parameters (5.Mixing)
mixing_type broyden
mixing_beta 0.7
mixing_gg0 0
STRU文件
ATOMIC_SPECIES
Zr 91.224 Zr_ONCV_PBE-1.0.upf
LATTICE_CONSTANT
1.8897261258369282 // lattice scaling factor (Bohr)
LATTICE_VECTORS
3.2390000000 0.0000000000 0.0000000000
0.0000000000 3.2390000000 0.0000000000
0.0000000000 0.0000000000 5.1720000000
ATOMIC_POSITIONS
Direct //Cartesian or Direct coordinate.
Zr // Element type
0.0 // magnetism(Be careful: value 1.0 refers to 1.0 bohr mag, but not fully spin up !!!)
2 // number of atoms
0.00 0.00 0.00 0 0 0//the position of atoms and other parameter specify by key word
0.25 0.25 0.25 1 1 1
KPT文件
K_POINTS //keyword for start
0 //total number of k-point, 0' means generate automatically
Gamma //which kind of Monkhorst-Pack method, Gamma' or `MP'
4 4 4 0 0 0 //first three number: subdivisions along recpri. vectors
//last three number: shift of the mesh
稳定结构的计算方法:
Zr的基态结构:
按照教程的方法配置INPUT、STRU、KPT文件,然后运行,迭代100次。最终得到STRU_NOW.cif文件,得到Zr的稳定的晶格结构及原子坐标。
得到的文件如下:
data_none
_audit_creation_method generated by ABACUS
_cell_length_a 3.17226
_cell_length_b 3.17226
_cell_length_c 5.05728
_cell_angle_alpha 90
_cell_angle_beta 90
_cell_angle_gamma 113.326
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
Zr 0.298569 0.701431 0.25
Zr 0.701431 0.298569 0.75
与materials project网站上的Zr的结构相对比,在lattice parameters和final structure方面都比较相似,在晶体结构上也较为相似,认为其稳定结构是合理的。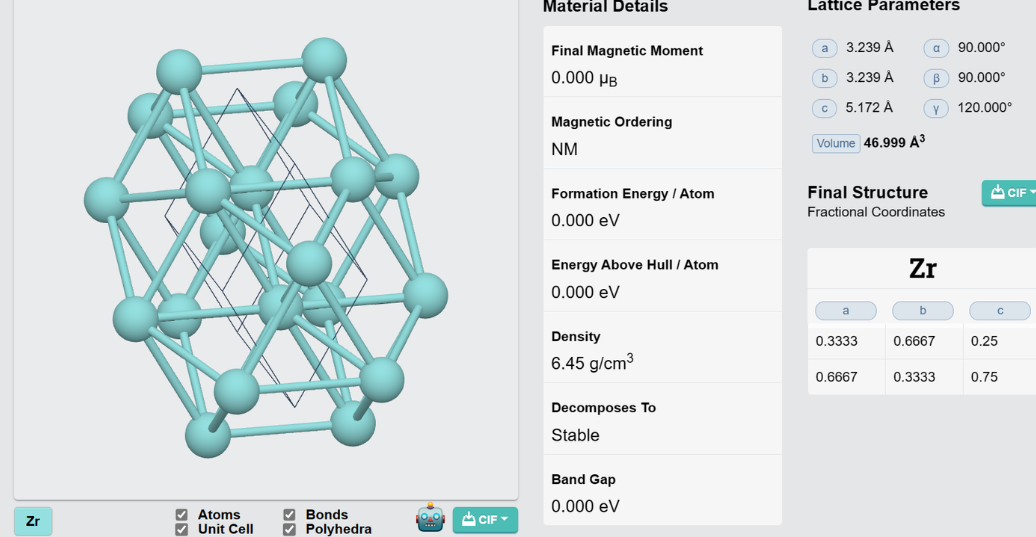
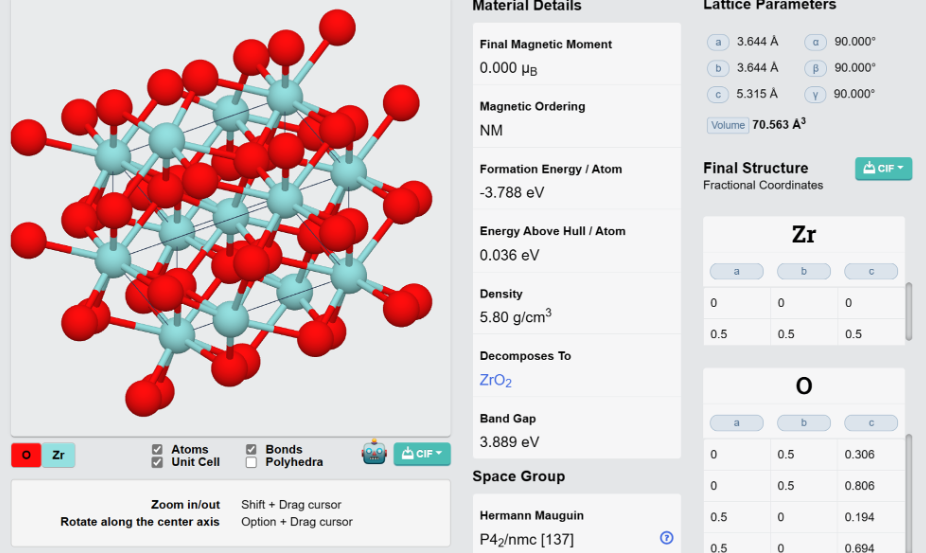
ZrO2的稳定结构:
选择锆的常见化合物,二氧化锆,配置STRU,INPUT,KPT文件,并进行迭代计算。得到结果如下图所示。 \
\
data_none
_audit_creation_method generated by ABACUS
_cell_length_a 3.60653
_cell_length_b 3.59601
_cell_length_c 5.27289
_cell_angle_alpha 90
_cell_angle_beta 90
_cell_angle_gamma 90
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
Zr 0 0 0.999038
Zr 0.5 0.5 0.500962
O 0 0.5 0.308833
O 0 0.5 0.807325
O 0.5 0 0.191167
O 0.5 0 0.692675
将其与实验值的ZrO2的结构对比,原子位置和长度等较接近。近似认为合理。