


蛋白质组学入门教程(0)数据预处理流程 · 基础篇
©️ Copyright 2023 @ Authors
作者: guolj@dp.tech 📨
日期: 2023-07-31
共享协议: 本作品采用知识共享署名-非商业性使用-相同方式共享 4.0 国际许可协议进行许可。
快速开始: 点击上方的 开始连接 按钮即可开始。
本文为 OpenMS 部分内容的搬运和魔改,主要面向希望做一些 LC-MS/MS 下机数据预处理,但又不知道从何开始的零基础同学。
如果你已经具备一定的基础,且主要关注于应用侧,你应该考虑开始接触 NextFlow,例如 QuantMS
规律,开始总是少数人认识,后来才是多数人认识。就是对少数人说来,也是从不认识到认识,也要经过实践和学习的过程。任何人开始总是不懂的,从来也没有什么先知先觉。
——〈读苏联《政治经济学教科书》的谈话〉,1959年12月至1960年2月
LC-MS/MS数据基础
当做完LC-MS/MS实验后,你一般可以导出一个原始文件,文件格式与你使用的仪器厂商息息相关,一些常见的例子可以见下表(来源参考ProteoWizard):
| 仪器厂商 | 下机原始文件格式 |
|---|---|
| ABI | T2D |
| Agilent | MassHunter .d |
| Bruker | Compass .d, YEP, BAF, FID, TDF |
| Sciex | WIFF / WIFF2 |
| Shimadzu | LCD |
| Thermo Scientific | RAW |
| Waters | MassLynx .raw / UNIFI |
利用ProteoWizard-msConvert、ThermoRawFileParser等工具,你可以将这些原始格式转变为数据预处理过程中常用的.mzML格式。
当然,除了上述提到的格式,还有其他非常多如今并不那么常用的格式,更为详细的介绍你可以查看wikipedia中的描述。
你可以在自己的终端中运行下面的代码安装依赖,不过在本Notebook中这并不必要
pip install pyopenms pandas openpyxl tqdm
同时,我们保留了每个cell中必要的import和变量定义,以便你不重新开机的情况下不必从头重新运行代码
接下来,让我们看看mzML中包含的数据是什么样的
以上便是一个LC-MS/MS实验下机数据中包含的所有主要数据,我们后续的一系列数据分析将以它们为基础展开。
你可以新开notebook的cell,然后逐个查看这些数据框的样子哦~就像下面这样:
原始数据预处理
数据格式转换
前述已经提过了数据格式转换的常用工具,你唯一需要注意的是,如果选择做PeakPicking,你将会获得centroided格式的数据;反之,你将获得profile模式的数据。
在这里,让我们节省一些时间,暂时跳过这个部分。当然,我会放一些示例代码在下面。
# 利用aspera从iprox下载数据
username=YourUserName # 你需要亲自去注册一个账号
ascp -d -QT -l1000m -P 33001 --file-manifest=text -k 2 -o Overwrite=diff ${username}@download.iprox.org:IPX0000755000 .
# 一、ThermoRaw格式转换为mzML
# 安装 thermorawfileparser
conda install -c bioconda thermorawfileparser
# 示例:转换为profile模式的mzML文件
ThermoRawFileParser -i=./IPX0000755000/IPX0000755003/ZY_UPS2_A1_Mouse_0_6ug.raw -f=2 -p=1- -o=./IPX0000755000/IPX0000755003/mzml_profile
# 示例:转换为centroid模式的mzML文件(当前一般情况下的推荐模式)
ThermoRawFileParser -i=./IPX0000755000/IPX0000755003/ZY_UPS2_A1_Mouse_0_6ug.raw -f=2 -p=1- -o=./IPX0000755000/IPX0000755003/mzml_centroid
# 二、ProteoWizard的msConvert(推荐)
# 首先我会推荐你使用Windows系统下ProteoWizard的GUI工具,因为部分仪器厂商的格式转换依赖部分dll文件,这当前只能在Windows系统下进行
# 然后,你可以利用docker+wine的方式
# https://hub.docker.com/r/chambm/pwiz-skyline-i-agree-to-the-vendor-licenses
docker pull chambm/pwiz-skyline-i-agree-to-the-vendor-licenses
docker run -it --rm -e WINEDEBUG=-all -v /your/data:/data chambm/pwiz-skyline-i-agree-to-the-vendor-licenses wine msconvert /data/file.raw
特征提取(Feature Detection)
在蛋白组学数据分析的预处理流程中,特征提取是一项重要的处理步骤。
特征提取的目标是从原始的质谱图谱数据中提取出具有生物学意义的特征,这些特征可以代表蛋白质或肽段。
具体来说,特征提取主要包括以下几个方面的处理:
- 在质谱全谱图中识别肽聚集特定质荷比下的特征峰。这些特征峰代表具有相近质荷比的一系列肽聚集。
- 拟合特征峰的同位素模式。根据含碳原子个数不同的肽聚集所形成的同位素峰进行拟合,得到特征的质荷比。
- 构建特征在经过色谱分离后在保留时间维度上形成的洗脱峰。合并不同扫描时刻的同一特征,获得其经整个色谱过程的洗脱规律。
- 计算各特征的积分强度。该强度反映特征物质的相对含量。
- 统计各特征的其他信息,如电荷状态、维度(质荷比和保留时间)、质量等。
特征提取将复杂的原始图谱转化为具有生物学意义且容易解析的特征矩阵,为后续的识别鉴定、定量分析等奠定基础。它是蛋白组学分析流程的关键步骤之一。
简单地说,在这个步骤中,我们将相近质荷比(一般高分辨率容许误差为5 ppm)以及相近保留时间的峰当作同一个特征(实际上指同一个分子,例如同一个肽段)。
OpenMS中有多个工具可以在LC-MS数据中识别这些特征,这些工具称为“FeatureFinder”。当前在pyOpenMS中可用的FeatureFinder有:
FeatureFinderMultiplexAlgorithm(例如SILAC、二甲基标记、非标记的肽特征检测)FeatureFinderAlgorithmPicked(非标记的肽特征检测)FeatureFinderIdentificationAlgorithm(非标记的、基于识别的肽特征检测)FeatureFinderAlgorithmIsotopeWavelet(老式仪器)FeatureFindingMetabo(非标记的、基于识别的代谢特征检测)FeatureFinderAlgorithmMetaboIdent(非标记的、基于识别的代谢特征检测)
除了FeatureFindingMetabo和FeatureFinderMetaboIdentCompound用于代谢组学数据和小分子之外,上述所有算法都是用于蛋白组学数据。
峰对齐(Map Alignment)
在做完特征识别后,我们还需要做峰对齐。它的基本原理如下:
- 对每个特征峰,计算其在不同样品之间在保留时间维度上的错位误差。
- 通过动态编程算法找到一种峰匹配方案,使得总的错位误差最小化。
- 根据峰匹配结果,用样条插值的方法对所有样品的特征峰在保留时间上进行一一对应的拟合,实现峰的对齐。
- 对对齐后的特征矩阵,在质荷比维度上进行去噪和归一化。
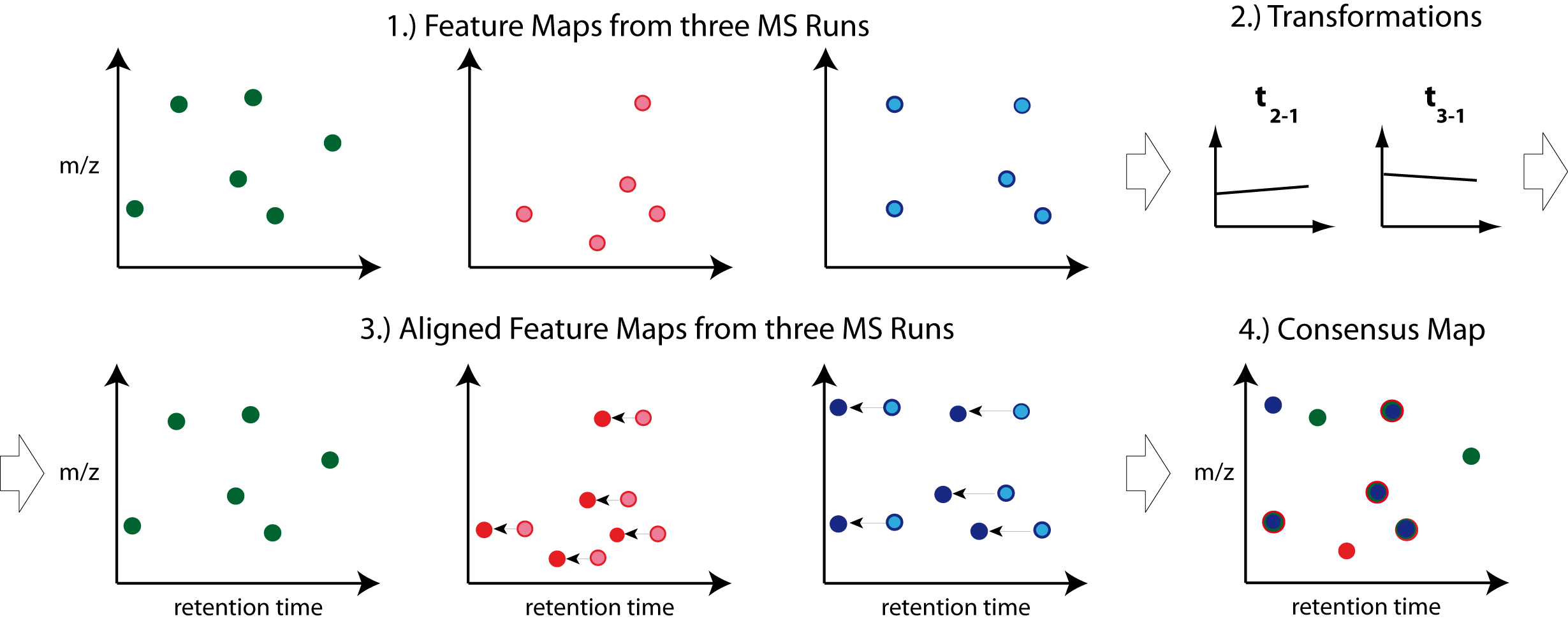
——图片来源pyOpenMS
通过峰对齐算法,可以消除不同测量中非生物学因素导致的特征峰位置偏差,使下游的定量分析更加准确可靠。
更多信息可以查阅OpenMS官方文档
在这里,我们采用MapAlignmentAlgorithmPoseClustering。
它是一种基于姿态聚类的图谱对齐算法[1]。
该算法为每个图谱选择 intensities 最高的 x 个峰/特征,通过分析峰对距离来找到保留时间最可能的转换。
在OpenMS中,这通过参数“max_num_peaks_considered”来模型化,它反过来影响运行时间和结果的稳定性。
更大的值会延长计算时间,更小的值可能导致没有或不稳定的转换矩阵。设置为-1使用所有特征(对大图谱会花很长时间)。

特征组合(Feature Linking)
做完特征提取和对齐后,我们便可以基于空间邻近关系进行(例如相似的保留时间和m/z)特征组合。
(可选地)我们也可以先对各个峰做基于二级谱的鉴定,再来做组合。这取决于你对速度和精度的偏好。
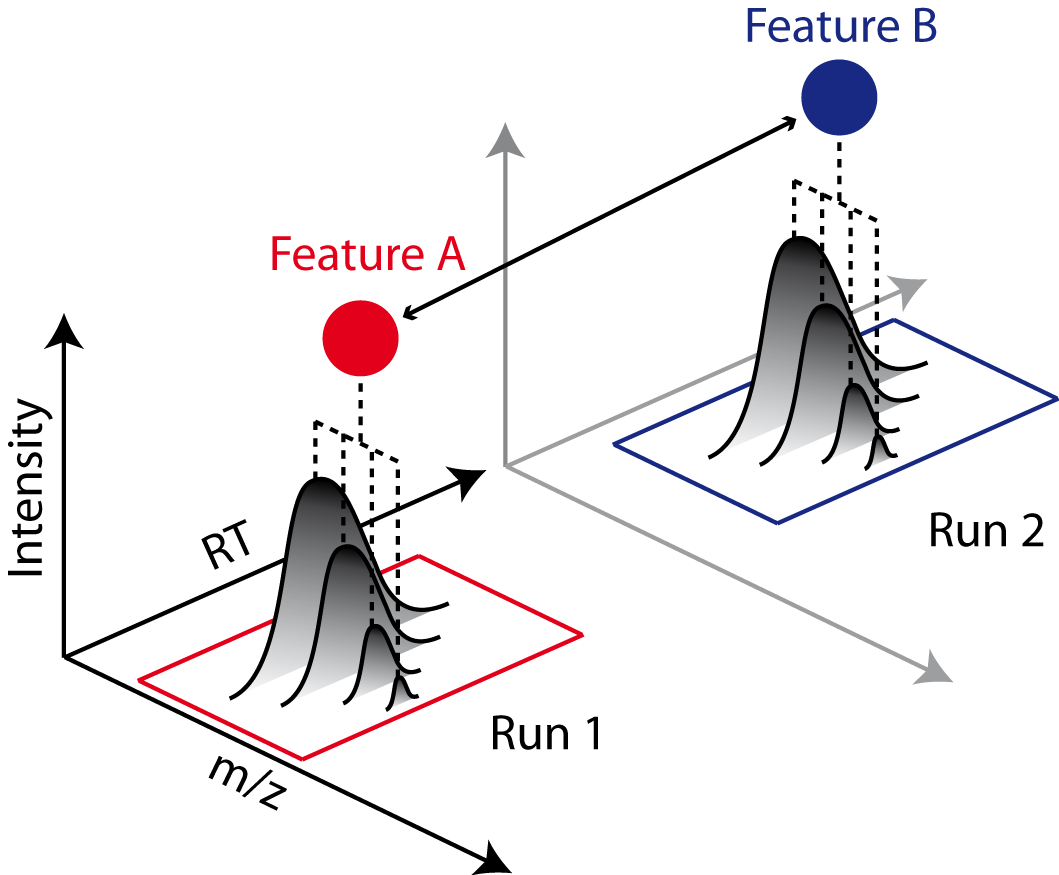
——图片来源pyOpenMS
pyOpenMS中提供了实现和运行特性略有不同的各种特征组合算法:
FeatureGroupingAlgorithmQTFeatureGroupingAlgorithmKDFeatureGroupingAlgorithmFeatureGroupingAlgorithmLabeledFeatureGroupingAlgorithmUnlabeled
你可以在OpenMS官方文档里找到更为详细的介绍。
接下来,我们采用FeatureGroupingAlgorithmQT进行特征组合。它是一个用于非标记数据的特征分组算法。
该算法取多个特征图或聚类图,并在不同图之间搜索对应的(聚类)特征。这些图必须进行对齐(即纠正保留时间畸变,例如使用某个MapAlignmentAlgorithm),但可以容忍小的偏差。
这个特定算法会累积所有输入图的特征,然后应用QT聚类的变体来找到对应的特征组。更多细节请参阅QTClusterFinder。
现在,你获得了一个ConsensusMap,它包含每个id对应的肽段的所有特征和丰度。
但是我们仍然不知道这些肽段的序列,更不要说获得各个蛋白在各样本中的含量,接下来,让我们来补全这个部分。
肽段识别(Peptide Search)
在基于质谱的蛋白组学中,碎片离子质谱(MS2)通常是通过将其与从FASTA数据库生成的一组理论质谱进行比较来解释的。
蛋白质组学中用于评价肽段识别结果质量的常用评分算法类型通常包括:
1、截断评分法
基于匹配结果的总体评分进行筛选,设定评分阈值,高于阈值的結果被保留。
评分通常依据匹配片段的强度求和计算。
简单直接,但可能过滤真阳性或保留假阳性。
2、概率评分法
基于匹配的概率模型打分。
通过比较匹配和随机匹配的概率分布差异给出评分。
更可靠,但需建立概率模型。
3、机器学习评分法
使用分类模型对匹配进行评分。
可以整合多维信息,训练出一个复杂的评分函数。
准确度高,但需要大量训练数据。
4、多级评分结合
结合上述多种评分方法,综合其优势。
例如先用概率评分进行筛选,再用机器学习进行排序。
可以使结果更可靠,但算法复杂度也更高。
在大多数蛋白组学应用中,会使用专用的搜索引擎来搜索数据。(如Comet、Crux、Mascot、MSGFPlus、MSFragger、Myrimatch、OMSSA、SpectraST或XTandem。它们的结果读取pyOpenMS都支持)
在这里,我们将使用OpenMS中的SimpleSearchEngineAlgorithm,仅用于教学示范。这使得将一个mzML文件与一个蛋白序列FASTA数据库进行搜索变得非常简单。
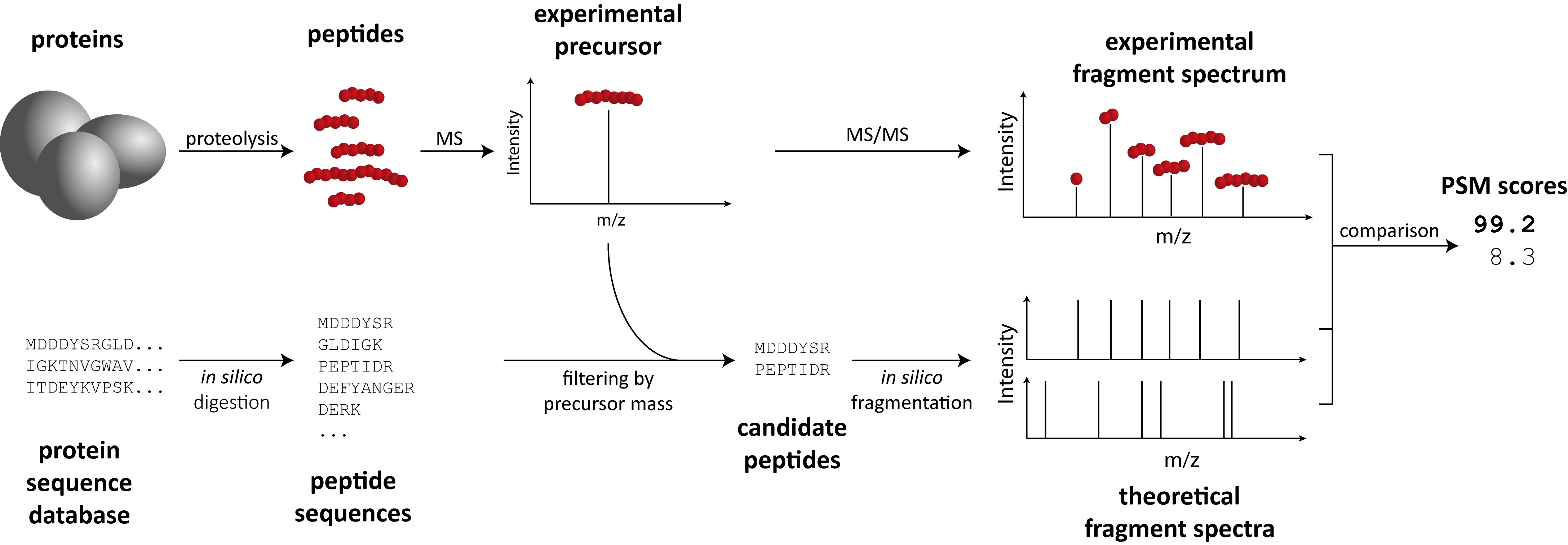
——图片来源pyOpenMS
你可以在OpenMS官方文档里找到更为详细的介绍。
如果你对各个搜索引擎的使用方式、各自的速度和精度等比较比较感兴趣,不妨留言催更。
由于每种方法都需要做评分,再根据评分决定一个特征应对的肽段。这时候便会涉及到假阳性的问题,它由以下几个原因造成:
存在可能的肽段数量太多,包含许多实际不存在的成分。
评分算法不够准确和可靠,导致一些低质量匹配获得较高分数。
真正存在的肽段因数据问题评分较低,被排除在结果之外。
多个结果整合时出现错误传递。
存在未知化合物的干扰。
为了提高结果的可靠性,除了需要不断优化算法和流程,我们还需要并利用对照组评估假阳性概率。
但由于我们大部分场景下并不存在可以用来评估假阳性率的对照组,于是便凭空造一个现实中极大概率并不存在的蛋白序列库——即decoy库,一般有序列反转法和序列打乱法。
根据搜索结果中decoy的命中情况,评估实际结果中假阳性的概率。这为后续判断结果的可靠性提供依据。
搜库结果映射(IDMapping)
接下来,我们把搜库的结果映射到之前对齐过的特征图上
这种映射是基于保留时间和质荷比进行的。
简单来说,如果一个肽鉴定的位置在一个共识特征的边界内或足够接近共识特征的质心,则将该肽鉴定分配给这个共识特征。
那些没能匹配到的肽鉴定仍然会以“未分配肽”的形式记录在结果图中。蛋白鉴定则会批注到整个结果图上,即不针对任何特定的共识特征。
在所有情况下,RT和m/z维度上的误差都是根据参数rt_tolerance和mz_tolerance应用的。这里的误差理解为“正负x”,所以实际的匹配范围是误差值的两倍。
如果有多个特征或共识特征的位置与某肽鉴定重叠(考虑到允许的误差),则该鉴定会被批注到所有这些特征上。
蛋白质定量
到目前为止,我们获得了各条肽段及其对应蛋白质在各个样品中的含量。那如何确定各蛋白质在样品中的含量呢?
在适合本数据的LFQ(Label-Free Quantitation)方法中,最常见地,我们会采用两种比较单纯的方法——TOP3和iBAQ。
其中TOP3如其名,每条蛋白我们取其中含量最高的三条肽段,取其平均作为蛋白质的定量结果。这个实现很简单,你不妨自己试试。
下次教程我们将展开讲解各个蛋白质定量方法,你可以通过点赞或留言的形式催更。



