


ABACUS基于平面波进行的收敛性测试与晶格弛豫(以Kr为例)
致谢:
原notebook链接:https://bohrium.dp.tech/notebooks/82328491785
Kr介绍
发现过程
在与瑞利共同发现氩气(1894年)之后,威廉∙拉姆齐(William Ramsay)与助手莫里斯∙特拉弗斯(Morris W. Travers)一起,继续寻找比氩气更轻的惰性气体家族的成员.1898年5月30日,从一升液态空气蒸馏出氧气、氮气、氩气之后,他们在剩下的液体中发现了这种新的惰性气体的光谱——独特的亮黄色和亮绿色线条暗示了这种一种新的元素,他们用希腊语κρυπτος将其命名为Krypton,并根据密度放在周期表的溴和铷之间.因为发现氪等多种稀有气体,拉姆齐获得了1904年的诺贝尔化学奖.
物理化学性质
常温下为无色气体,在高压电管中呈现白色和蓝色光芒;固体氪是一种白色结晶物质,具有面心立方结构.原子序数为36,相对原子质量为83.798,属于稀有气体(惰性气体),在元素周期表上位于VIIIA族(18族),第四周期;电子排布为[Ar]3 4IA族(18族),第四周期;电子排布为[Ar]3 4
DFT算法介绍
DFT(Density Functional Theory)即密度泛函理论,基于量子力学理论,以电子密度为基本变量,将能量表示为电子密度的泛函,从而求解多粒子体系性质,是物理计算材料学和计算化学领域的常用计算方法.
在求解定态薛定谔方程时,哈密顿算符包括了电子动能、质子动能、电子与电子、质子与质子、电子与质子的相互作用共五项.若直接求解由N个电子M个质子组成的系统,其包含个变量,会使求解变得十分困难,因此,进行一些适当的近似尤为重要.
Hohenberg-Kohn定理
Hohenberg-Kohn定理 1: 对任意相互作用的电子系统,处在外部势场V中,则该外部势场V可由基态电子密度唯一决定(除了相差一个常数)
Hohenberg-Kohn定理 2: 对任一给定的外势,可定义关于电子密度的普适性泛函,其中,基态(非简并)电子密度使得此能量泛函取极小值
1994年提出的Hohenberg-Kohn定理大大降低了求解自由度(降低到3),奠定了密度泛函的基础.该定理表明,多电子系统的性质全部由电子系统的哈密顿量决定,但是,Hohenberg-Kohn定理并未给出电子密度泛函的具体形式.
Kohn-Sham方程
Kohn-Sham假设(KS假设):有相互作用系统的基态电荷密度,可以被一个无相互作用系统的基态电荷密度表示出来
KS方程 其中,第一项是电子动能,第二项是外势,第三项是Hartree势(电子-电子相互作用),第四项是交换关联势.后三项被称为有效势
至此,将求解有相互作用的系统变为求解无相互作用的系统(用单体波函数描述电子),而相互作用的部分归入交换关联部分.
交换关联势求解
常见的求解交换关联势的方式有:局域密度近似(LDA)、广义梯度近似(GGA)、含动能密度的广义梯度近似(meta-GGA)、杂化泛函.
自洽计算过程
给定初始电荷密度,构造哈密顿量,求解KS方程,计算新的电荷密度并判断是否收敛,如果不收敛则重复2-4步直到电荷密度收敛(自洽)
ABACUS计算方法介绍
ABACUS是基于密度泛函理论(DFT)的第一性原理计算软件,通过电子自洽迭代(SCF)计算获得电子结构的总能量、能级、电子波函数、电子密度等关键信息.
输入文件介绍
ABACUS的三个基本输入文件是 STRU、KPT、INPUT .其中STRU 文件提供基本结构信息;KPT 文件提供周期性边界条件下布里渊区采样的网格设置;INPUT 文件提供计算所需的各种设置参数.
STRU文件
本文Kr的赝势文件选择为download/pseudopotentials/nc-sr-05_pbe_standard_upf/Kr.upf
ATOMIC_SPECIES
Kr 83.798 Kr.upf
NUMERICAL_ORBITAL
#此处写轨道文件
LATTICE_CONSTANT
1.8897261258369282
LATTICE_VECTORS
5.8357000000 0.0000000000 0.0000000000
0.0000000000 5.8357000000 0.0000000000
0.0000000000 0.0000000000 5.8357000000
ATOMIC_POSITIONS
Direct
Kr
0.0000000000
4
0.0000000000 0.0000000000 0.0000000000 1 1 1 mag 0.0
0.5000000000 0.5000000000 0.0000000000 1 1 1 mag 0.0
0.5000000000 0.0000000000 0.5000000000 1 1 1 mag 0.0
0.0000000000 0.5000000000 0.5000000000 1 1 1 mag 0.0
第 2 行 为"元素 原子质量 赝势文件名称" .
第 5 行提供数值轨道文件名,如果采用 pw 基组计算,则不需要写第 4~5 行内容.
第 8 行代表晶格整体缩放的一个长度,表明接下来的坐标均以 Angstrom 为单位(1 Angstrom=1.8897261258369282 bohr).
第 11~13 行表示建立的坐标系x,y,z方向所对应的三条晶格矢量.
第 16 行 "Direct" 表示采用分数坐标(晶格坐标)
第 17 行表示下面的信息属于哪种原子,这里是Kr.
第 18 行设置原子初始磁矩,如果 INPUT 里的 nspin 参数设为 1,则不考虑磁性,这个参数不起作用.
第 19 行表示体系的Kr原子个数.
第 20~27 行给出所有 8 个Kr原子的坐标;前三个数是坐标,之后的(1 1 1)三个数分别代表允许该原子在x,y,z方向上移动;"mag 0.0" 指定每个原子的初始磁矩,若设置此参数,则覆盖第 18 行的值.
KPT文件
K_POINTS
0
Gamma
4 4 4 0 0 0
第二行表示k点是自动生成的,也可以自行设置总数.
第三行表示划分布里渊区网格的方法.
第四行前三个数为x,y,z方向的k点个数,后三个数表示三个方向上网格是否平移,这里表示不平移.
INPUT文件
INPUT_PARAMETERS
#Parameters (1.General)
suffix Kr,k=4
calculation scf
symmetry 1
pseudo_dir .
orbital_dir .
basis_type pw
ecutwfc 100
#Parameters (2. SCF iterations)
scf_nmax 20
scf_thr 1e-8
#Parameters (3. Solve KS equation)
nbands 26
ks_solver cg
#Parameters (4.Smearing)
smearing_method gauss
smearing_sigma 0.015
#Parameters (5.Mixing)
mixing_type broyden
mixing_beta 0.7
mixing_gg0 0
suffix 表示输出文件后缀.
calculation 表示计算方法,这里采用SCF.
symmetry 表示是否考虑对称性,1表示考虑对称性,0表示仅考虑时间反演对称性,-1是不考虑对称性.打开对称性可以减少计算时间.
orbital_dir、orbital_dir 分别是赝势文件和轨道文件的路径,"."表示在当前文件夹中.
basis_type 表示使用基组类型,这里使用平面波(pw).
ecutwfc 表示截止能量,单位是Ry,其大小决定了计算的精度.
scf_nmax 表示最大迭代次数,半导体和绝缘体都较容易收敛,因此这里取20.
scf_thr 是相邻电子迭代步之间电荷密度的误差,是判断是否收敛的标准.
nbands 计算公式为,其中是占据能级数,对于Kr,根据选择的赝势文件的信息,得到价电子为8,由于每个能级可以填充两个电子,共4个原子,因此,nbands为26.
ks_solve 表示展开哈密顿矩阵的对角化方法,这里使用pw基组下默认的cg.
之后的参数4和5分别是关于展宽方法和电荷密度混合的设置.
收敛性测试
使用脚本更改输入文件并提取各输入条件下OUT.suffix文件夹内的running_scf.log里的数据.
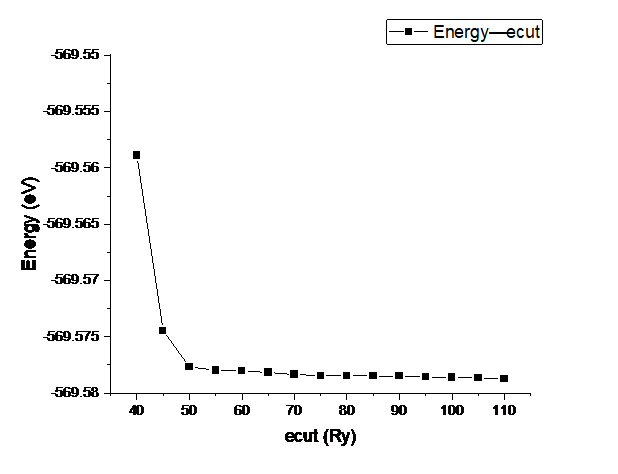
ecut测试
先对ecut进行测试,k暂时取4.发现在ecut=70Ry时收敛.
k点测试
观察曲线,k取4时能量收敛.4和5时间接近,所以选4.

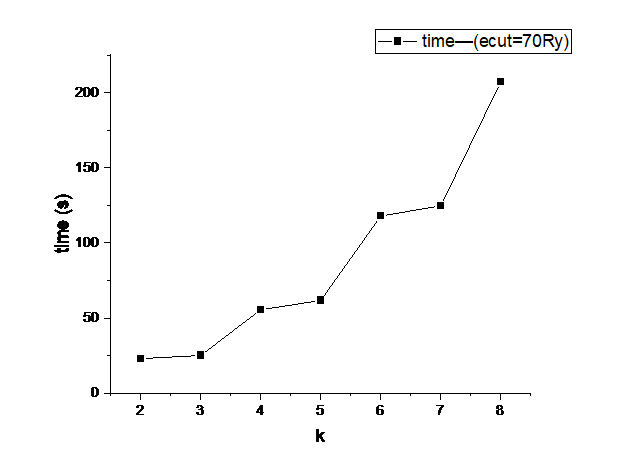
由于开启了对称性,所以实际计算的k点数量有了简化,这就是图中总点数不同但计算时间接近的原因.
Kr晶格弛豫
输入文件如下
INPUT
INPUT_PARAMETERS
suffix Kr
ntype 1
pseudo_dir .
ecutwfc 60
scf_thr 1e-4
basis_type pw
calculation cell-relax
force_thr_ev 0.01
stress_thr 2
relax_nmax 100
out_stru 1
STRU
ATOMIC_SPECIES
Kr 83.798 Kr.upf
LATTICE_CONSTANT
1.8897261258369282
LATTICE_VECTORS
5.8357000000 0.0000000000 0.0000000000
0.0000000000 5.8357000000 0.0000000000
0.0000000000 0.0000000000 5.8357000000
ATOMIC_POSITIONS
Direct
Kr
0.0000000000
4
0.0000000000 0.0000000000 0.0000000000 1 1 1
0.5000000000 0.5000000000 0.0000000000 1 1 1
0.5000000000 0.0000000000 0.5000000000 1 1 1
0.0000000000 0.5000000000 0.5000000000 1 1 1
KPT
K_POINTS
0
Gamma
4 4 4 0 0 0
运行
经过两次迭代就得到了结果. 输出为
data_none
_audit_creation_method generated by ABACUS
_cell_length_a 5.90294
_cell_length_b 5.90294
_cell_length_c 5.90294
_cell_angle_alpha 90
_cell_angle_beta 90
_cell_angle_gamma 90
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
Kr 0 0 0
Kr 0.5 0.5 0
Kr 0.5 0 0.5
Kr 0 0.5 0.5
与输入相比基本无变化,说明输入的已经是稳定结构.
结构优化
输入文件如下
INPUT
NPUT_PARAMETERS
suffix KrF2_relax
ntype 2
pseudo_dir .
ecutwfc 60
scf_thr 1e-4
basis_type pw
calculation cell-relax
force_thr_ev 0.1
stress_thr 6
relax_nmax 100
out_stru 1
STRU
F赝势文件选择download/pseudopotentials/nc-sr-05_pbe_standard_upf/F.upf
ATOMIC_SPECIES
Kr 83.798 Kr.upf
F 18.998 F.upf
LATTICE_CONSTANT
1.8897259886 # 1.8897259886 Bohr = 1.0 Angstrom
LATTICE_VECTORS
4.73200 0.00000 0.00000
0.00000 4.73200 0.00000
0.00000 0.00000 6.03700
ATOMIC_POSITIONS
Direct
Kr
0.0
2
0.0 0.0 0.0 1 1 1
0.5 0.5 0.5 1 1 1
F
0.0
4
0.2106 0.7894 0.5 1 1 1
0.2894 0.2894 0.0 1 1 1
0.7106 0.7106 0.0 1 1 1
0.7894 0.2106 0.5 1 1 1
KPT
K_POINTS
0
Gamma
4 4 4 0 0 0
运行
经过12次迭代,运行结果为
data_none
_audit_creation_method generated by ABACUS
_cell_length_a 4.47063
_cell_length_b 4.47063
_cell_length_c 5.27534
_cell_angle_alpha 90
_cell_angle_beta 90
_cell_angle_gamma 90
loop_
_atom_site_label
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
Kr 0 0 0
Kr 0.5 0.5 0.5
F 0.203514 0.796486 0.5
F 0.296486 0.296486 0
F 0.703514 0.703514 0
F 0.796486 0.203514 0.5
仅晶格长度和位置发生微小变化,说明初始就比较稳定了.
晶格的形状与网站上的 mp-558928类似.
INPUT的stress_thr以及force_thr_ev原本没有设置成这么大,但是观察迭代过程,发现10次之后没有下降趋势,所以认为继续迭代下去的意义不是很大,重新设置了INPUT参数.
稳定晶格结构如图所示.
