


pyGenomeTracks:多维基因组数据的高质量可视化

嘿!你是否曾经为了分析和可视化基因组数据而感到头疼?面对大量的复杂信息,你是否感觉有点不知所措?别担心,我来帮你解决这个问题!
让我向你介绍pyGenomeTracks(PGT),这是一个令人兴奋的新软件,它可以帮助你在基因组数据的海洋中找到自己的方向。你可能会问,这个软件有什么特别之处?好问题!
首先,我们都知道基因组数据分析涉及许多复杂的步骤,而PGT正是为了解决这些挑战而诞生的。它可以轻松处理大规模数据,帮助你在全基因组水平上进行快速而高效的分析和总结。对于研究人员来说,这意味着更多的时间和精力可以用于深入挖掘数据中隐藏的宝藏。
其次,PGT还提供了强大的可视化功能,可以让你在基因组上绘制各种数据轨迹。无论是基因注释、基因表达还是染色质信号和互作信息,它都能轻松应对。最重要的是,它可以将这些信息融合到一个统一的图像中,让你一目了然。
PGT的使用也非常简单!你只需要准备一个配置文件,指定你想要绘制的轨迹和数据源,然后运行一个简单的命令行,就能生成高质量的图像。不用再为复杂的数据处理过程和图像生成而烦恼了!
如果你喜欢图形界面,也别担心!PGT还提供了图形化界面,让你可以更直观地进行操作。
所以,如果你是一个对基因组数据感兴趣的研究者或生物信息学家,PGT绝对是你不可或缺的利器!它将帮助你轻松解决数据分析和可视化难题,让你在基因组探索的旅程中事半功倍!
让我们一起走进pyGenomeTracks的神奇世界,开启一段令人激动的基因组探索之旅吧!
📖 上手指南
共享协议:本作品采用知识共享署名-非商业性使用-相同方式共享 4.0 国际许可协议进行许可。
快速开始:本文档可在 Bohrium Notebook 上直接运行。点击上方的 开始连接 按钮,选择 ubuntu22-py310-r43-gpu-0803n:plot镜像 、R kernel和任意节点即可开始。
Basic Examples
A minimal example of a configuration file with a single bigwig track looks like this:
usage: pyGenomeTracks --tracks tracks.ini --region chr1:1000000-4000000 -o image.png pyGenomeTracks: error: argument --tracks: can't open './examples/bigwig_track.ini': [Errno 2] No such file or directory: './examples/bigwig_track.ini'
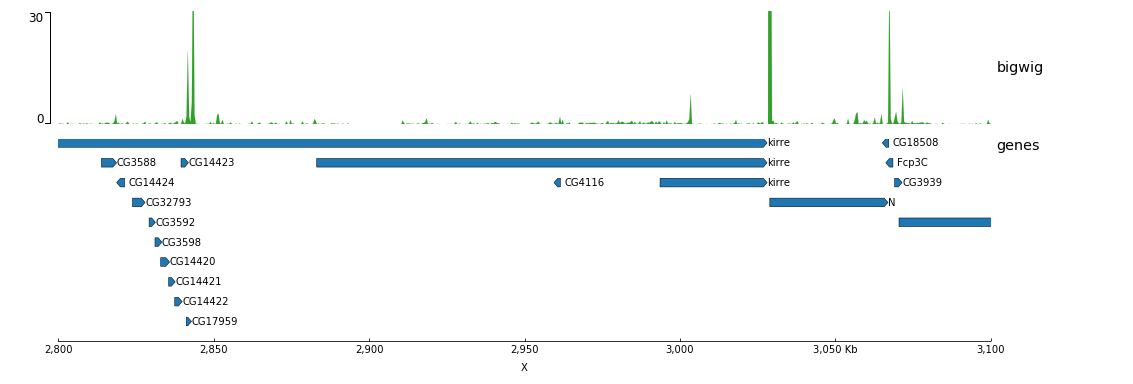
Now, let’s add the genomic location and some genes:
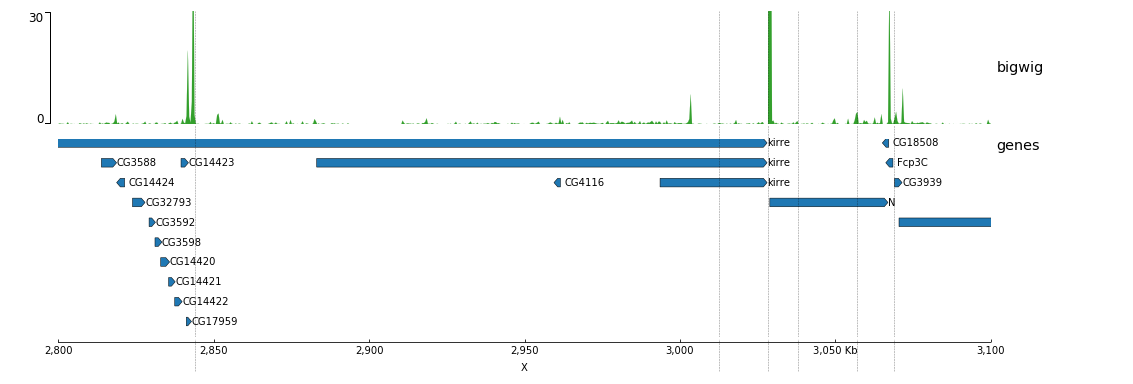
Now, we will add some vertical lines across all tracks. The vertical lines should be in a bed format.
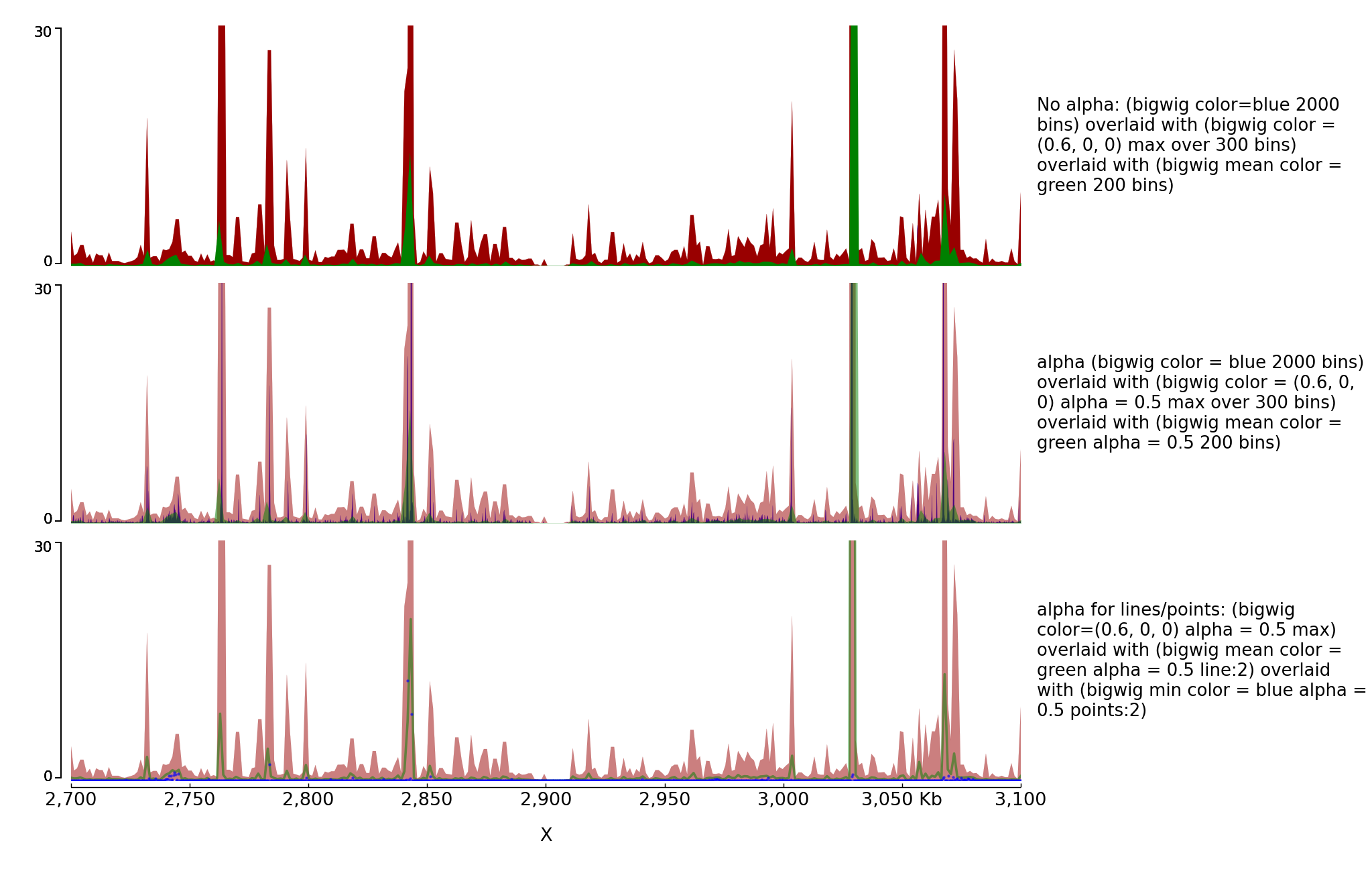
You can also overlay bigwig with or without transparency.
Examples with bed and gtf
Here is an example to explain the parameters for bed and gtf:
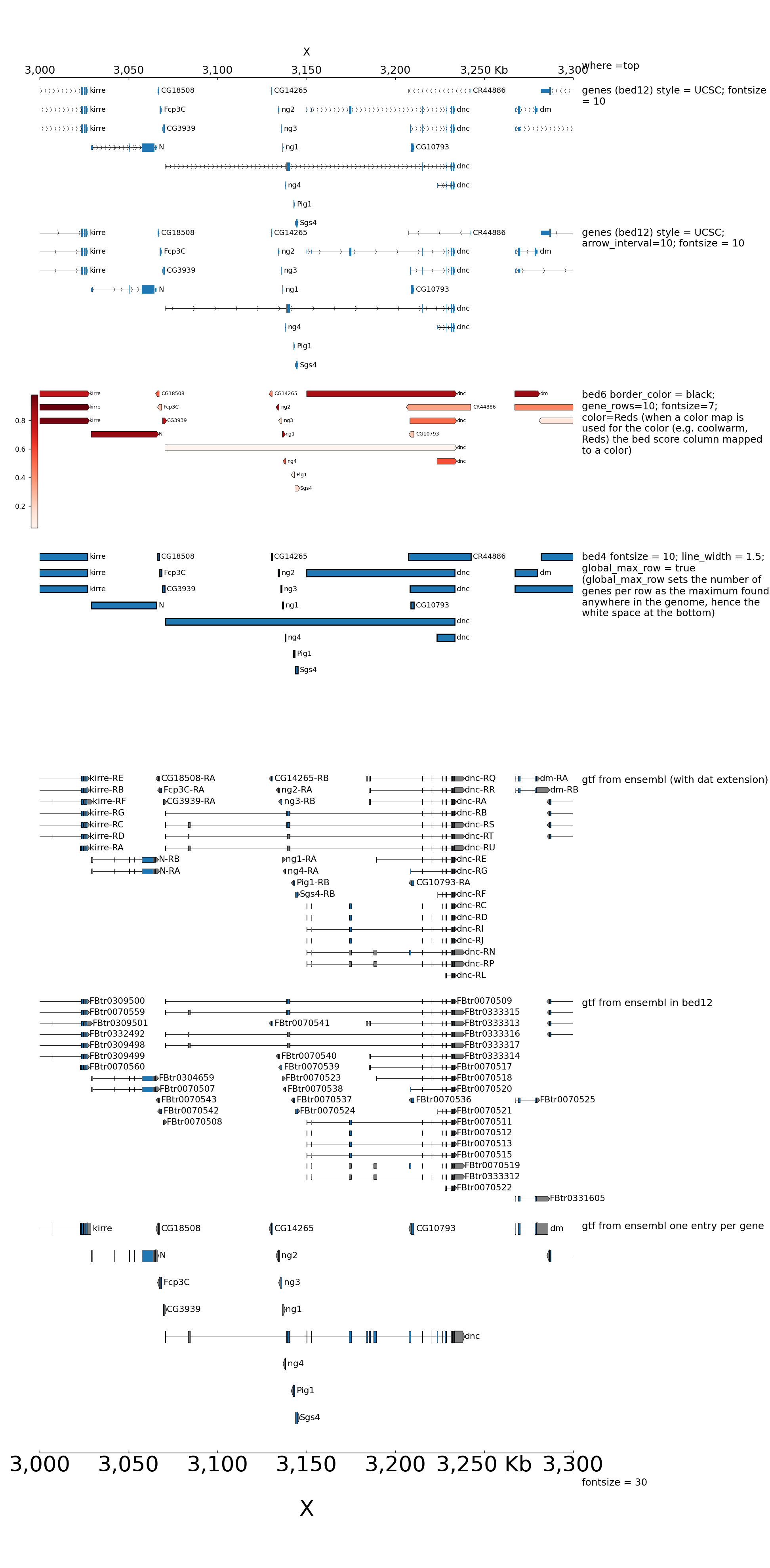
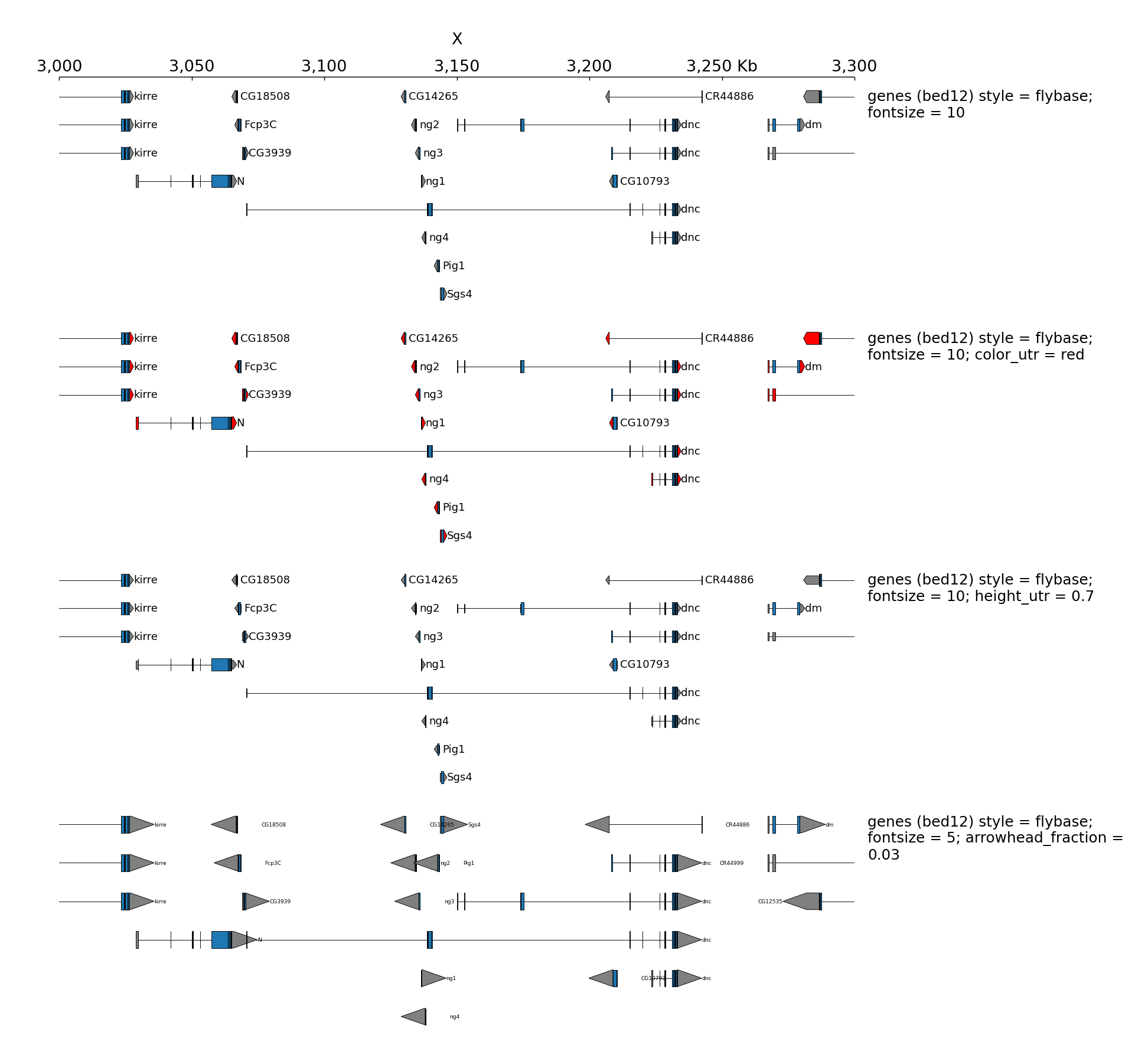
By default, when bed are displayed and interval are stranded, the arrowhead which indicates the direction is plotted outside of the interval. Here is an example to show how to put it inside:
![]()
When genes are displayed with the default style (flybase), the color and the height of UTR can be set:
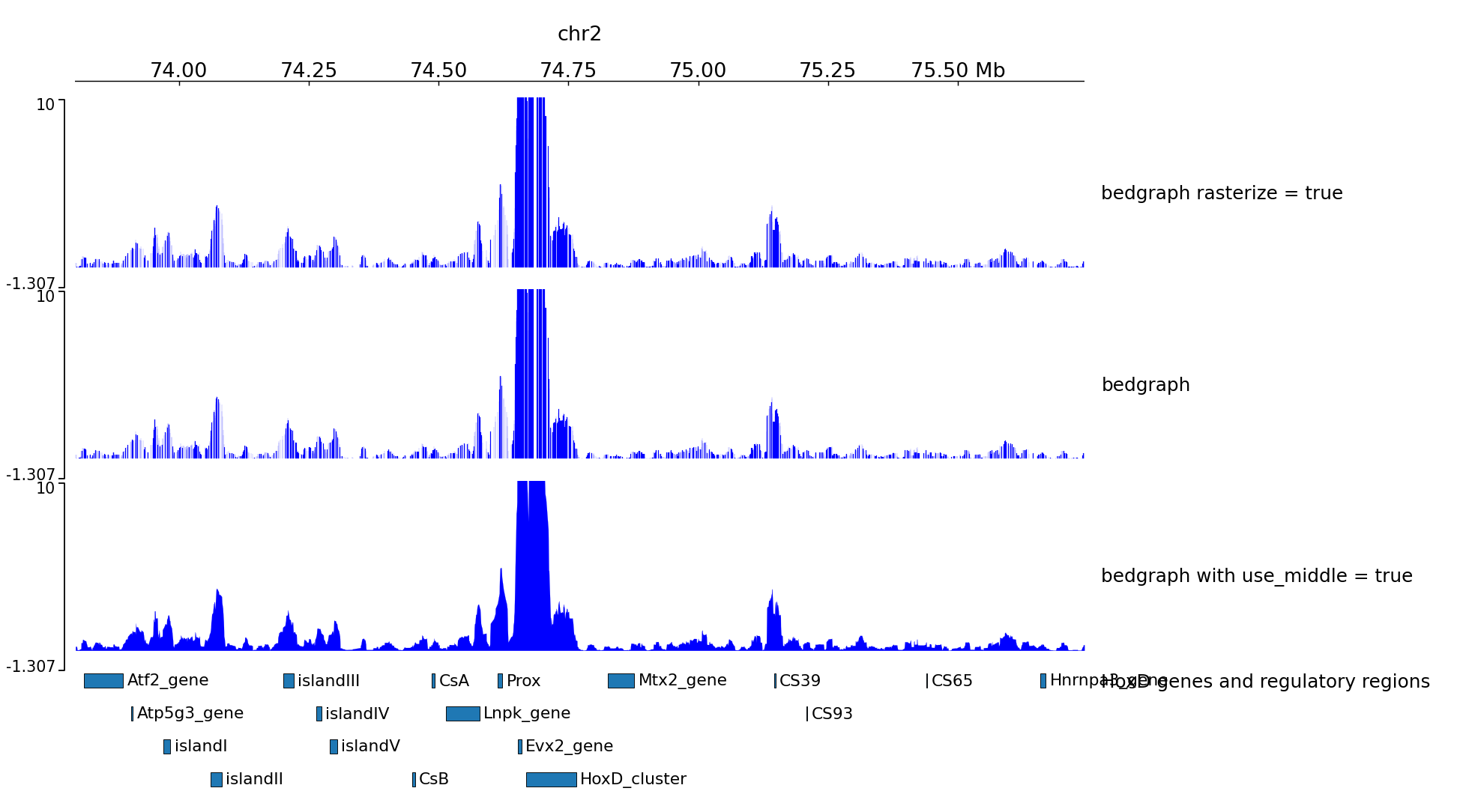
Examples with 4C-seq
The output file of some 4C-seq pipeline are bedgraph where the coordinates are the coordinates of the fragment. In these cases, it can be interesting to remove the regions absent from the file and just link the middle of the fragments together instead of plotting a rectangle for each fragment. Here is an example of the option use_middle.
We can generate two zooms using a bed instead of regions:
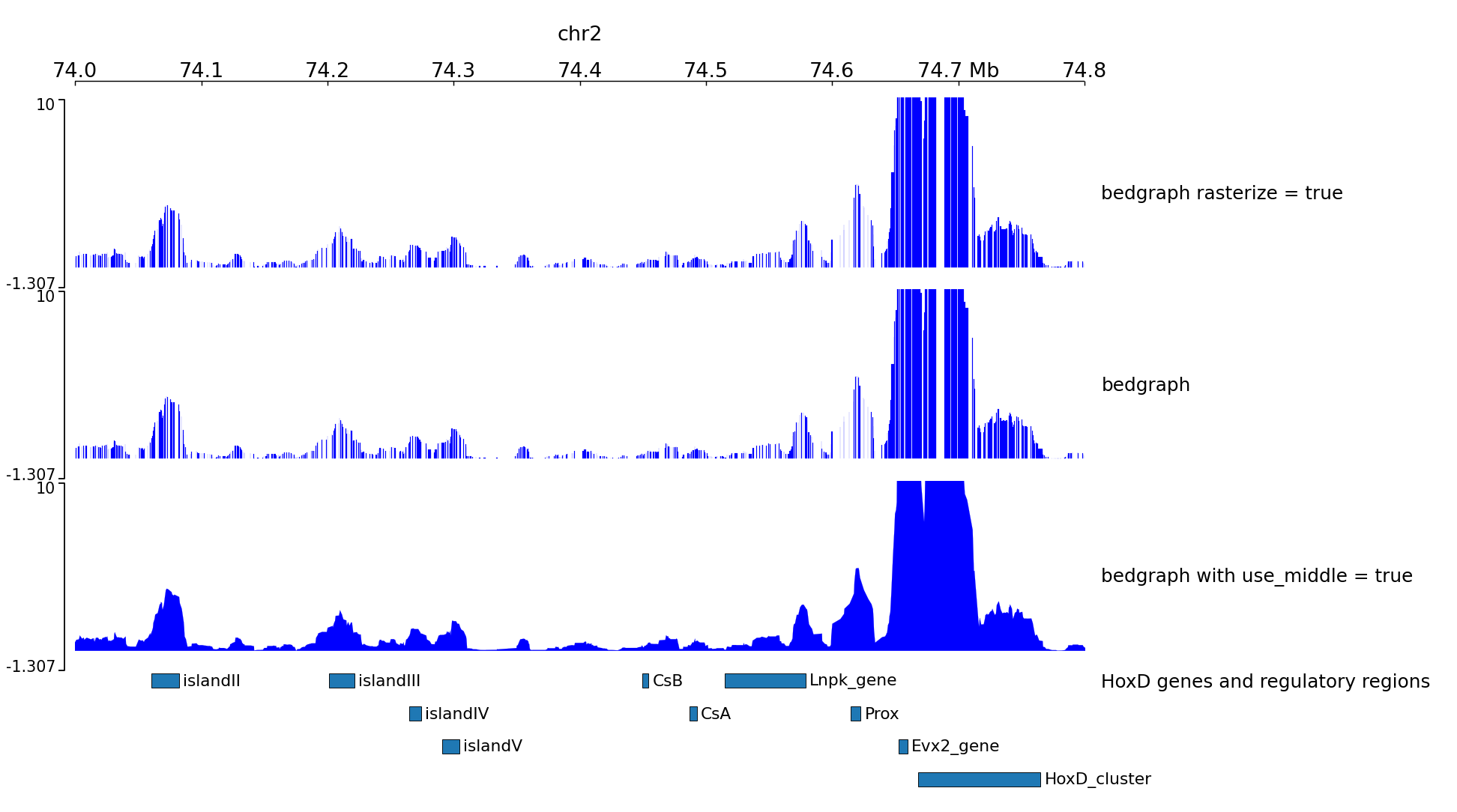
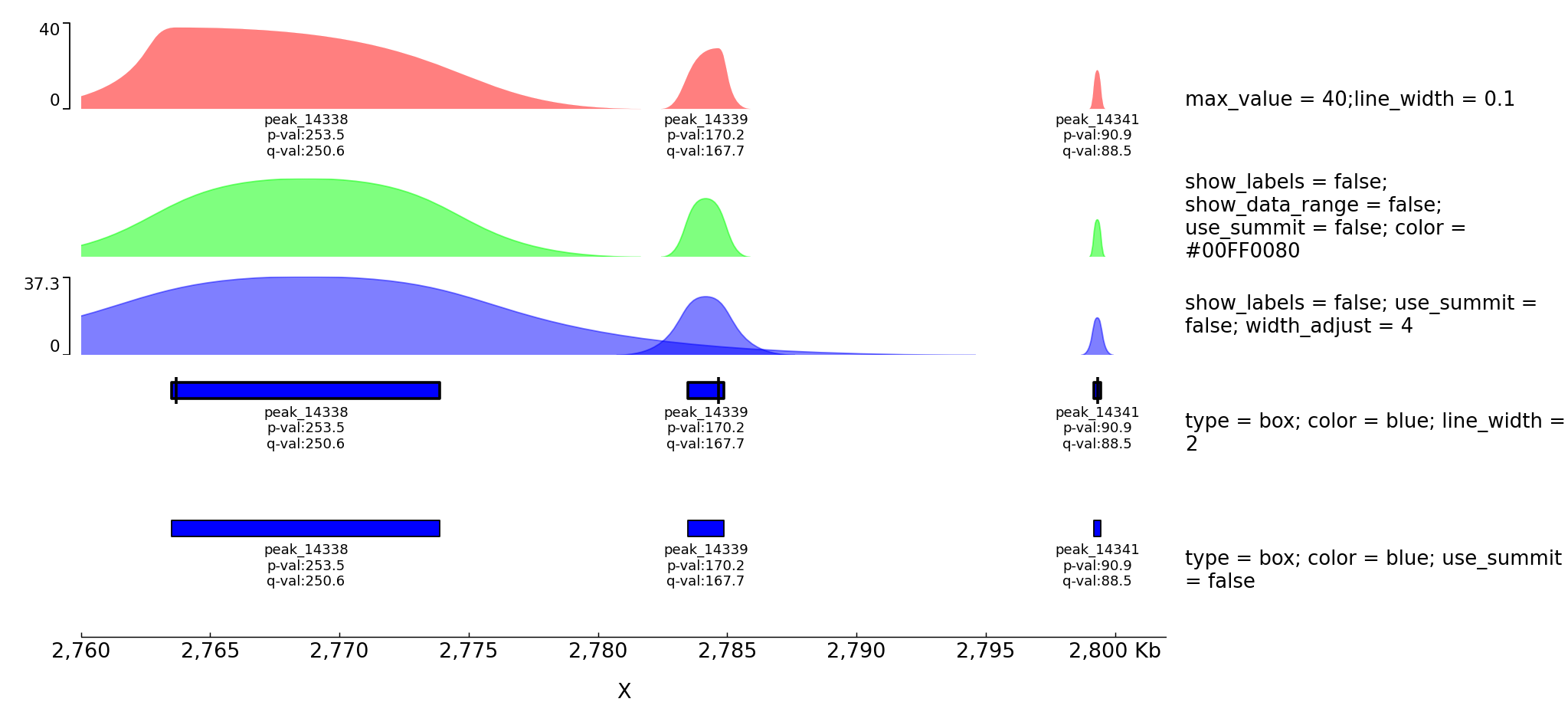
Examples with peaks
pyGenomeTracks has an option to plot peaks using MACS2 narrowPeak format.
The following is an example of the output in which the peak shape is drawn based on the start, end, summit and height of the peak.
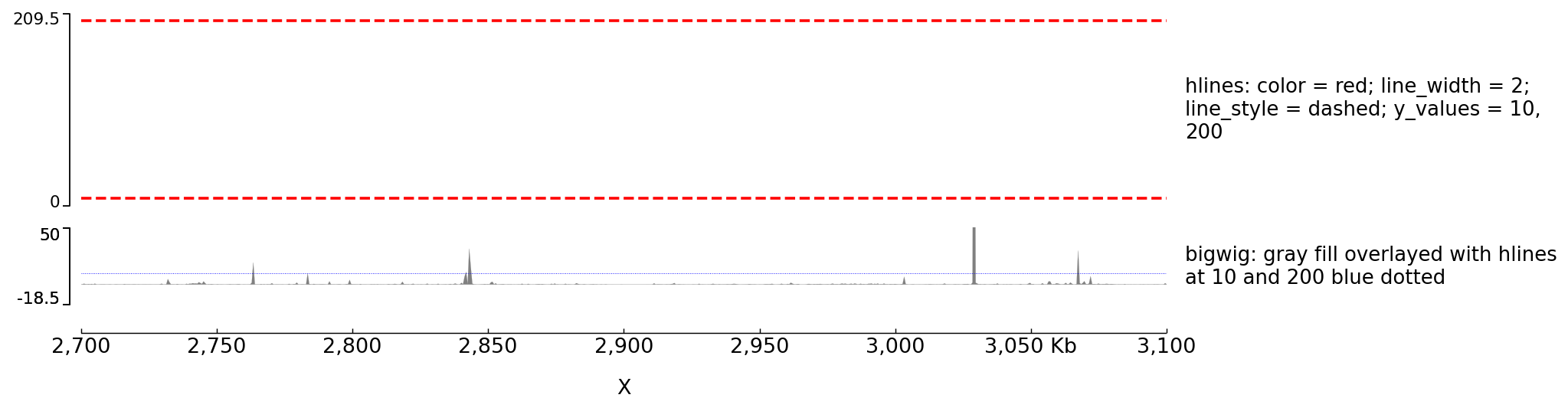
Example with horizontal lines
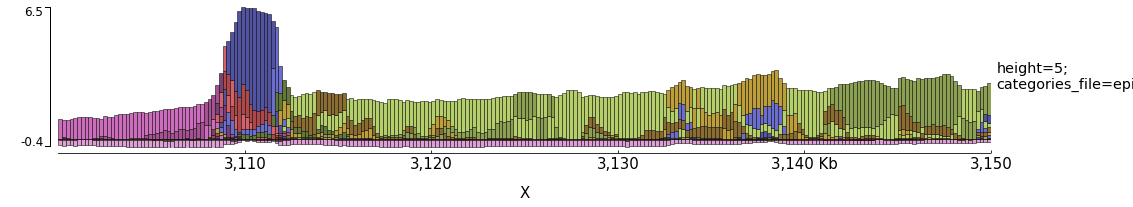
Examples with Epilogos
pyGenomeTracks can be used to visualize epigenetic states (for example from chromHMM) as epilogos. For more information see: https://epilogos.altiusinstitute.org/
To plot epilogos a qcat file is needed. This file can be crated using the epilogos software (https://github.com/Altius/epilogos).
An example track file for epilogos looks like:
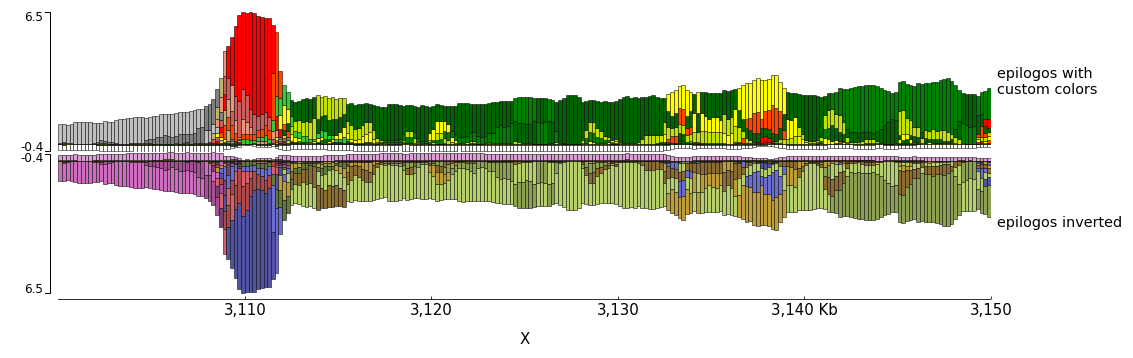
The color of the bars can be set by using a json file. The structure of the file is like this:
In the following examples the top epilogo has the custom colors and the one below is shown inverted.
Examples with multiple options
A comprehensive example of pyGenomeTracks can be found as part of our automatic testing. Note, that pyGenomeTracks also allows the combination of multiple tracks into one using the parameter: overlay_previous = yes or overlay_previous = share-y. In the second option the y-axis of the tracks that overlays is the same as the track being overlay. Multiple tracks can be overlay together.

The configuration file for this image is:
Examples with multiple options for bigwig tracks
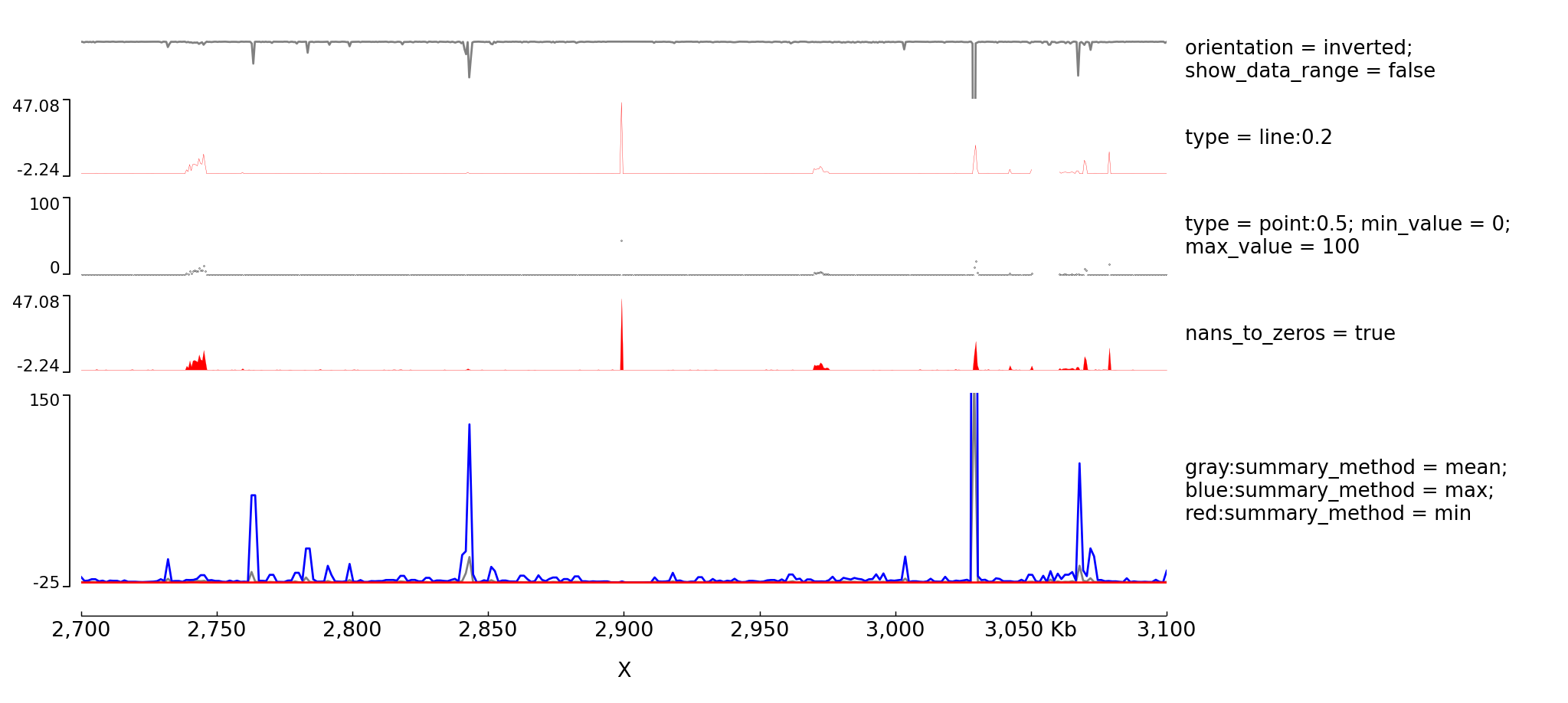 The configuration file for this image is:
The configuration file for this image is:
Examples with Hi-C data
The following is an example with Hi-C data overlay with topologically associating domains (TADs) and a bigwig file.
Here is an example where the height was set or not set and the heatmap was rasterized (default) or not rasterized (the dpi was set very low just to show the impact of the parameter).
The output is available here: master_plot_hic_rasterize_height.pdf.
This examples is where the overlay tracks are more useful. Notice that any track can be overlay over a Hi-C matrix. Most useful is to overlay TADs or to overlay links using the triangles option that will point in the Hi-C matrix the pixel with the link contact. When overlaying links and TADs is useful to set overlay_previous=share-y such that the two tracks match the positions. This is not required when overlying other type of data like a bigwig file that has a different y-scale.
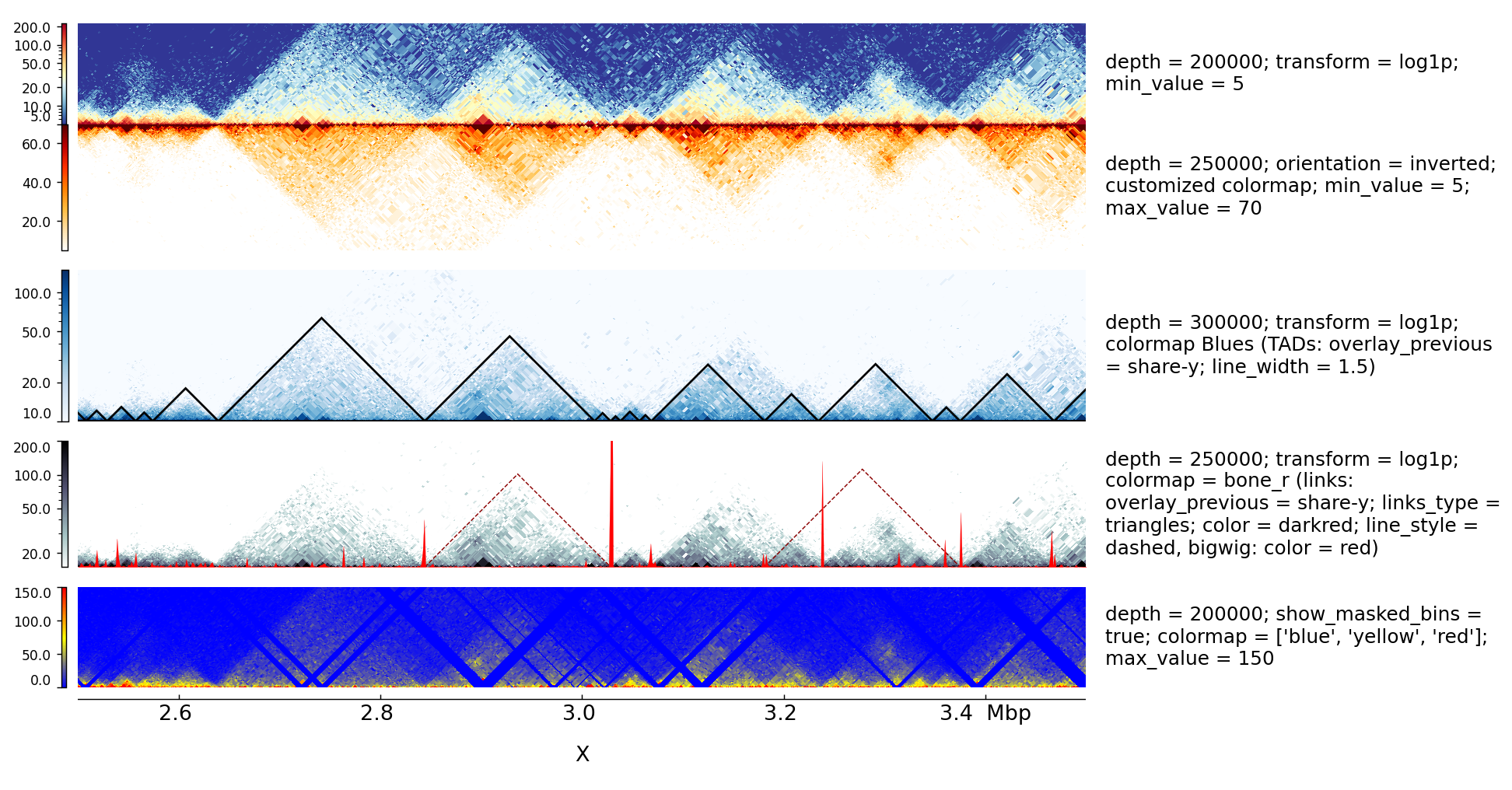
The configuration file for this image is:
Log transform and Operation Examples
With the parameter operation you can make operations between one or two files (here two bigwig files but this is also working with two bedgraph files). For example, difference, log ratio, scaling…
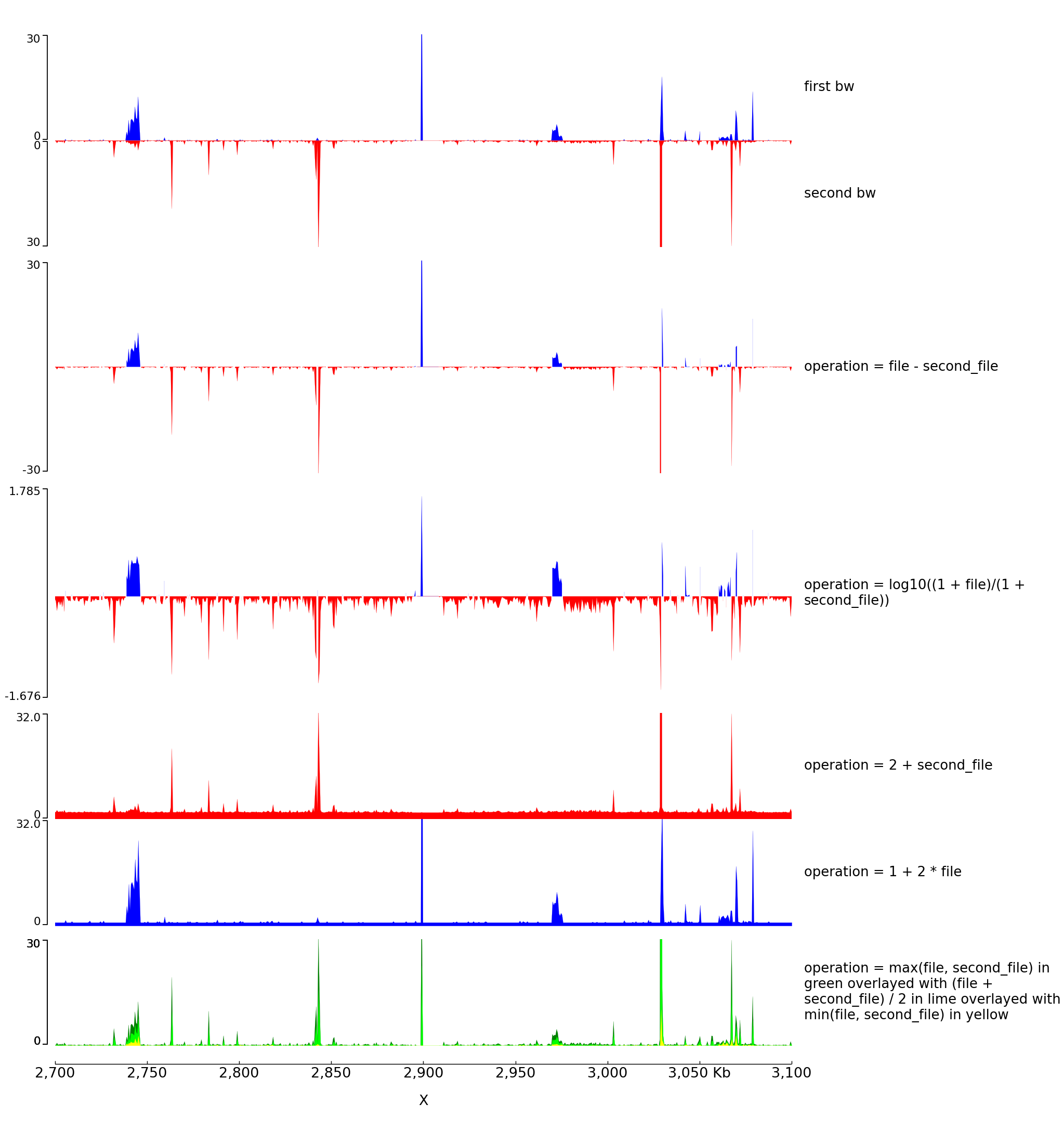
The configuration file for this image is:
With the parameter transformation you can log transform your data and decide to put on the y axis either the transformed values or the original values:
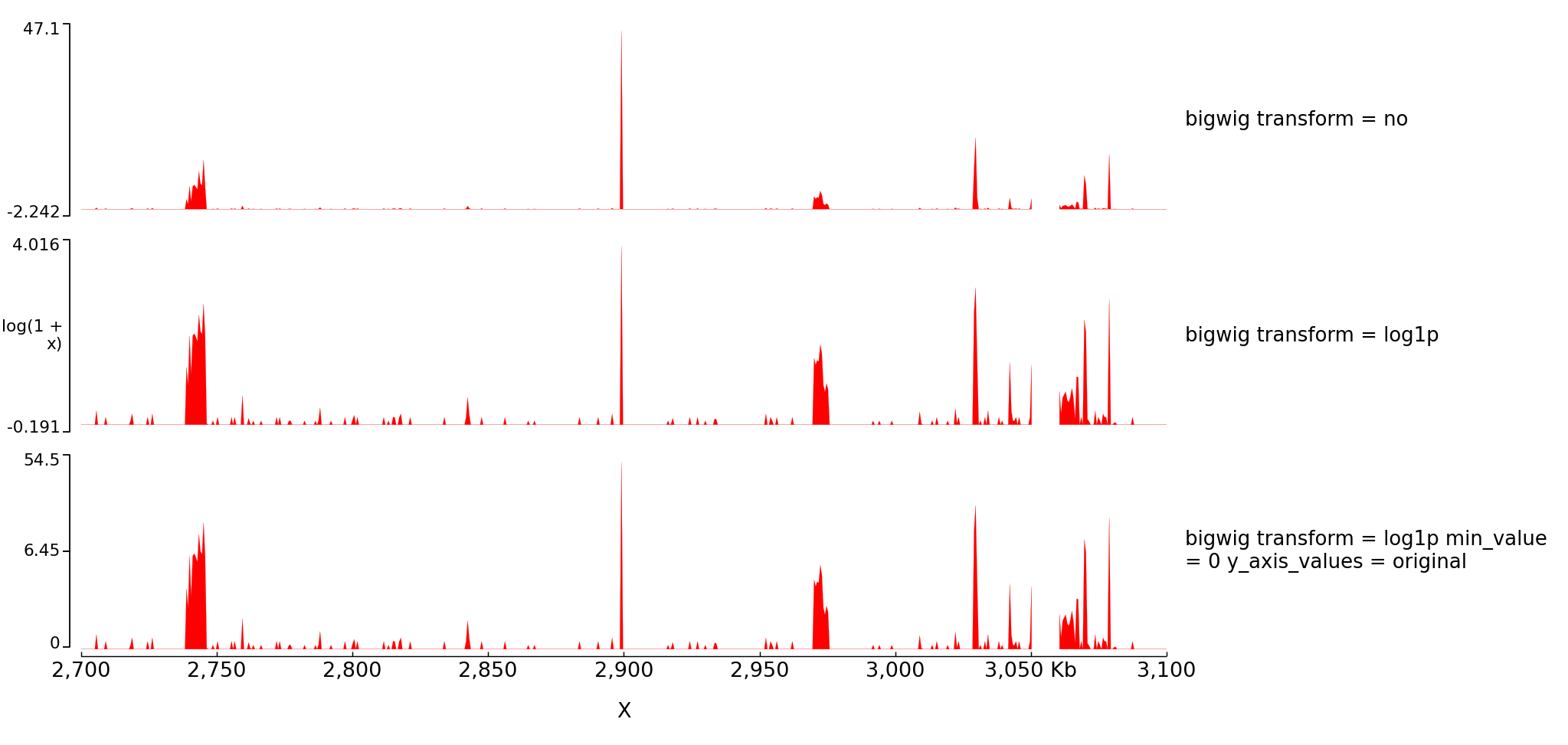
The configuration file for this image is:
With operation you can also do log transformation however nothing will be written on the left of the y axis:
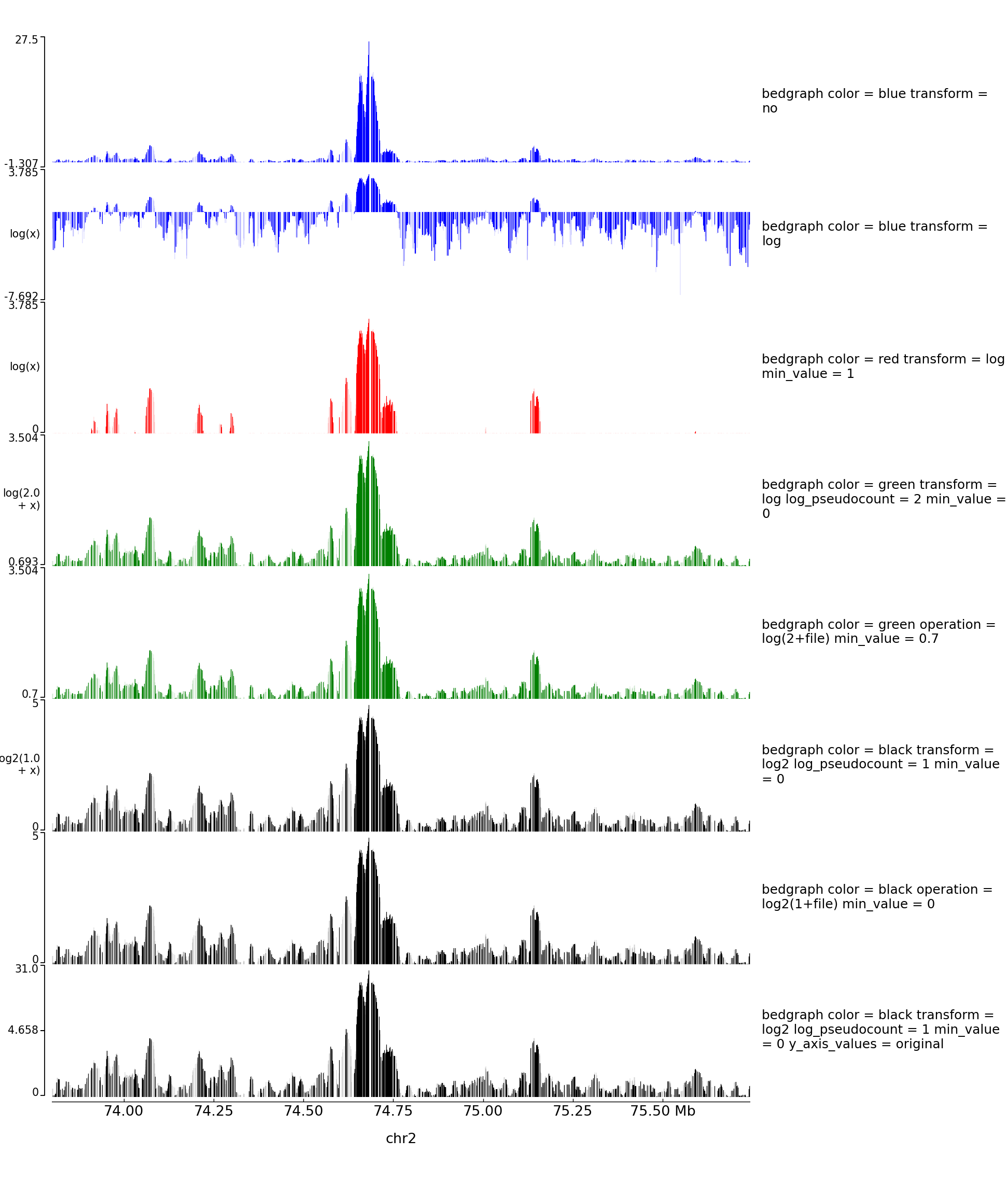
The configuration file for this image is:
References
- Lopez-Delisle L, Rabbani L, Wolff J, Bhardwaj V, Backofen R, Grüning B, Ramírez F, Manke T. pyGenomeTracks: reproducible plots for multivariate genomic data sets. Bioinformatics. 2020 Aug 3:btaa692. doi: 10.1093/bioinformatics/btaa692. Epub ahead of print. PMID: 32745185.
