


实验介绍
本教程的目的是帮助大家学习如何使用LAMMPS进行分子动力学(MD)模拟。本实验将模拟碳纳米管(CNT)在拉伸应力下的断裂过程,并分析其力学行为。
为了说明经典力场和反作用力场之间的区别,本教程分为两部分。在第一部分,使用了经典的力场,并且 CNT 原子之间的键是牢不可破的。在第二部分,使用了一个反作用力场(称为 AIREBO ),当 CNT 发生强烈变形时,化学键会断裂。

为什么有些模拟中,键不会发生断裂❓
在大多数经典的分子动力学力场中,原子之间的化学键是在仿真开始时设置的。无论在仿真过程中施加到原子上的力如何,键都保持完整。相邻原子之间的键通常由具有给定平衡距离的弹簧组成 和一个常数 : .此外,通常应用角度和二面体约束来保持相邻原子的相对方向。
第一部分: 建模
打开 VMD,转到 Extensions、Modeling,然后转到 Nanotube Builder。一个名为碳纳米结构的窗口打开,允许我们选择生成由石墨烯或氮化硼 (BN) 制成的片材或纳米管。在本教程中,我们生成一个碳纳米管。保留所有默认值,然后单击 Generate Nanotube(生成单壁碳管)。
在这一点上,这不是分子动力学模拟,而是一团未连接的点。在 VMD 终端中,通过在 VMD 终端中键入以下命令来设置框尺寸:
molinfo top set a 80
molinfo top set b 80
molinfo top set c 80
周期性盒子在每个方向的值设置为 80,因此盒子比碳纳米管大得多。要生成初始 LAMMPS 的data文档,我们将使用VMD的 Topotools:在 VMD 终端中输入以下命令生成 LAMMPS data文件:
topo writelammpsdata cnt_molecular.data molecular #这里的molecular 代表lammps的data文件的类型,cnt_molecular.data代表文件的名称
Notes: Topotools 可以从原子的相应位置推断出键、角、二面体和异体的位置,并生成一个 LAMMPS 可以读取的 .data 文档 [19]。有关 Topotools 的更多详细信息,请访问 Axel Kohlmeyer 的个人页面。
约束的参数(键长、二面体系数等)将在后面给出。一个名为 cnt_molecular.data 的新文档已经创建,它的开头是这样的:
700 atoms
1035 bonds
2040 angles
4030 dihedrals
670 impropers
1 atom types
1 bond types
1 angle types
1 dihedral types
1 improper types
-40.000000 40.000000 xlo xhi
-40.000000 40.000000 ylo yhi
-12.130411 67.869589 zlo zhi
(...)
cnt_molecular.data 文档包含有关碳原子位置的信息,以及由键、角度、二面体和 impropers 约束连接的原子的标识。将 cnt_molecular.data 文档保存在名为 unbreakable-bonds/ 的文档夹中。
Lammps的输入(input)文件
在 unbreakable-bonds/ 中创建一个新的文本文档,并将其命名为 input.lammps。将以下行复制到其中:
variable T equal 300
units real # 这里定义系统使用的单位类型为:real
atom_style molecular # 这里定义data文件的类型为:molecular
boundary f f f # 由于盒子的尺寸远大于CNT,所以边界条件是固定的
pair_style lj/cut 14 # LJ势的截止距离
bond_style harmonic
angle_style harmonic
dihedral_style opls
improper_style harmonic
special_bonds lj 0.0 0.0 0.5
read_data cnt_molecular.data
所选的单位系统是实数(因此距离以埃为单位,时间以飞秒为单位),atom_style 是分子(因此原子是可以相互结合的点),边界条件是固定的。边界条件在这里并不重要,因为盒子边界远离 CNT。
就像在 Lennard-Jones 流体中一样,对样式是 lj/cut(即具有短程截止的 Lennard-Jones 势),参数为 14,这意味着只有彼此距离小于 14 埃的原子才能通过 Lennard-Jones 势相互作用。
bond_style、angle_style、dihedral_style 和 improper_style 命令指定用于限制原子相对位置的不同势。有关此处使用的势的更多详细信息,您可以查看 LAMMPS 网站,例如参见 OPLS 二面体样式的页面 [20]。
最后一个命令 read_data 导入先前用 VMD 生成的 cnt_molecular.data 文档,其中包含有关盒子大小、原子位置等的信息。
通过键连接的原子通常不通过 Lennard-Jones 相互作用。因此,必须将有界原子排除在 Lennard-Jones 势计算之外。这里,这是通过 special_bonds 命令完成的。special_bonds 命令的三个数字是通过键连接的原子(分别为直接结合、由两个键分隔和由三个键分隔之间的 Lennard-Jones 相互作用的加权因子。例如,第一个加权因子的值为 0,它规定通过键连接的两个原子不通过 Lennard-Jones 势相互作用(因此它们仅通过键合石墨烯原子的谐波势相互作用)。
我们需要指定键合和非键合电位的参数。这里,参数取自 OPLS-AA (Optimised Potentials for Liquid Simulations-All-Atom) 力场[21]。在 unbreakable-bonds/ 文档夹中创建一个新的文本文档,并将其命名为 parm.lammps。将以下行复制到其中:
-40.000000 40.000000 xlo xhi
-40.000000 40.000000 ylo yhi
-12.130411 67.869589 zlo zhi
让我们通过将以下行添加到 input.lammps 来重新定位 CNT:
group carbon_atoms type 1
variable carbon_xcm equal -1*xcm(carbon_atoms,x)
variable carbon_ycm equal -1*xcm(carbon_atoms,y)
variable carbon_zcm equal -1*xcm(carbon_atoms,z)
displace_atoms carbon_atoms move <span class="katex"><span class="katex-html" aria-hidden="true"><span class="base"><span class="strut" style="height:0.8444em;vertical-align:-0.15em;"></span><span class="mord"><span class="mord mathnormal">c</span><span class="mord mathnormal">a</span><span class="mord mathnormal" style="margin-right:0.02778em;">r</span><span class="mord mathnormal">b</span><span class="mord mathnormal">o</span><span class="mord"><span class="mord mathnormal">n</span><span class="msupsub"><span class="vlist-t vlist-t2"><span class="vlist-r"><span class="vlist" style="height:0.1514em;"><span style="top:-2.55em;margin-left:0em;margin-right:0.05em;"><span class="pstrut" style="height:2.7em;"></span><span class="sizing reset-size6 size3 mtight"><span class="mord mathnormal mtight">x</span></span></span></span><span class="vlist-s"></span></span><span class="vlist-r"><span class="vlist" style="height:0.15em;"><span></span></span></span></span></span></span><span class="mord mathnormal">c</span><span class="mord mathnormal">m</span></span></span></span></span>{carbon_ycm} ${carbon_zcm}
他的第一个命令包括一个名为 carbon_atoms 的组中类型 1 的所有原子(即这里的所有原子)。carbon_xcm、carbon_ycm 和 carbon_zcm 这 3 个变量分别用于测量组carbon_atoms沿所有 3 个方向的当前位置。然后,displace_atoms 命令将组carbon_atoms移动,确保其质心位于原点 (0, 0, 0)。
change_box all x final -40 40 y final -40 40 z final -40 40
Notes: 这种更清晰、更对称的初始状态可以简化未来的数据分析,但不会对分子动力学产生任何影响。
将在 CNT 的边缘施加位移。为此,让我们从两条边中分离出原子,并将它们分别放入名为 rtop 和 rbot 的组中。将以下行添加到 input.lammps 中:
variable zmax equal bound(carbon_atoms,zmax)-0.5
variable zmin equal bound(carbon_atoms,zmin)+0.5
region rtop block INF INF INF INF ${zmax} INF
region rbot block INF INF INF INF INF <span class="katex"><span class="katex-html" aria-hidden="true"><span class="base"><span class="strut" style="height:0.8889em;vertical-align:-0.1944em;"></span><span class="mord"><span class="mord mathnormal" style="margin-right:0.04398em;">z</span><span class="mord mathnormal">min</span></span><span class="mord mathnormal">re</span><span class="mord mathnormal" style="margin-right:0.03588em;">g</span><span class="mord mathnormal">i</span><span class="mord mathnormal">o</span><span class="mord mathnormal">n</span><span class="mord mathnormal" style="margin-right:0.02778em;">r</span><span class="mord mathnormal">mi</span><span class="mord mathnormal">d</span><span class="mord mathnormal">b</span><span class="mord mathnormal" style="margin-right:0.01968em;">l</span><span class="mord mathnormal">oc</span><span class="mord mathnormal" style="margin-right:0.03148em;">k</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span></span></span></span>{zmin} ${zmax}
该变量对应于最后一个原子沿负 0.5 Ångstroms 的坐标,以及第一个原子沿正 0.5 Ångstroms 的坐标。然后,定义三个区域,分别对应:, (rbot, 区域底部) (rmid, 区域 middle) 和 (rtop, 区域顶部)
最后,让我们通过添加到 input.lammps 来定义 3 组原子,分别对应于位于三个区域中每个区域的原子:
group carbon_top region rtop
group carbon_bot region rbot
group carbon_mid region rmid
在 carbon_top 和 carbon_bot 组中选择的边的原子可以用不同的颜色表示。
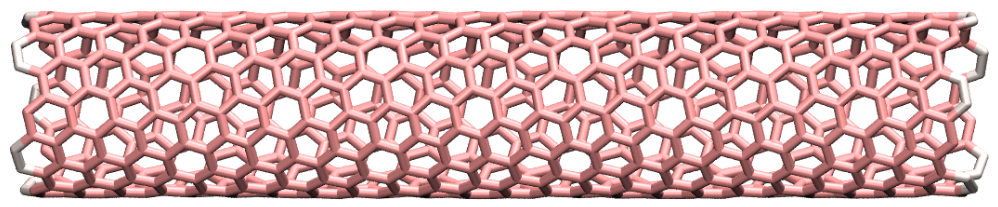
图:CNT,carbon_mid 基团的原子为粉红色,carbon_top 和 carbon_bot 基团的原子为白色。 运行仿真时,每组中的原子数打印在终端中(以及 log.lammps 文档中)。始终确保每组中的原子数与预期相对应,就像这里一样:
10 atoms in group carbon_top
10 atoms in group carbon_bot
680 atoms in group carbon_mid
最后,为了从一个不太理想的状态开始,创建一个具有一些缺陷的系统,让我们随机删除一小部分碳原子。为了避免删除太靠近边缘的原子,让我们定义一个新的区域名称 rdel,它从 CNT 边缘开始 Å。
variable zmax_del equal ${zmax}-2
variable zmin_del equal <span class="katex"><span class="katex-html" aria-hidden="true"><span class="base"><span class="strut" style="height:0.7429em;vertical-align:-0.0833em;"></span><span class="mord"><span class="mord mathnormal" style="margin-right:0.04398em;">z</span><span class="mord mathnormal">min</span></span><span class="mspace" style="margin-right:0.2222em;"></span><span class="mbin">+</span><span class="mspace" style="margin-right:0.2222em;"></span></span><span class="base"><span class="strut" style="height:0.8889em;vertical-align:-0.1944em;"></span><span class="mord">2</span><span class="mord mathnormal">re</span><span class="mord mathnormal" style="margin-right:0.03588em;">g</span><span class="mord mathnormal">i</span><span class="mord mathnormal">o</span><span class="mord mathnormal">n</span><span class="mord mathnormal" style="margin-right:0.02778em;">r</span><span class="mord mathnormal">d</span><span class="mord mathnormal">e</span><span class="mord mathnormal" style="margin-right:0.01968em;">l</span><span class="mord mathnormal">b</span><span class="mord mathnormal" style="margin-right:0.01968em;">l</span><span class="mord mathnormal">oc</span><span class="mord mathnormal" style="margin-right:0.03148em;">k</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord mathnormal" style="margin-right:0.13889em;">NF</span></span></span></span>{zmin_del} ${zmax_del}
group rdel region rdel
delete_atoms random fraction 0.02 no rdel NULL 482793 bond yes
delete_atoms 命令从 rdel 组中随机删除 2% 原子(即大约 10 个原子)。
The molecular dynamics
让我们指定系统的热化和动力学。将以下行添加到 input.lammps 中:
reset_atoms id sort yes
velocity carbon_mid create ${T} 48455 mom yes rot yes
fix mynve all nve
compute Tmid carbon_mid temp
fix myber carbon_mid temp/berendsen <span class="katex"><span class="katex-html" aria-hidden="true"><span class="base"><span class="strut" style="height:0.6833em;"></span><span class="mord"><span class="mord mathnormal" style="margin-right:0.13889em;">T</span></span></span></span></span>{T} 100
fix_modify myber temp Tmid
在使用 velocity 命令之前,必须重置 atom ID,这是通过 reset_atoms 命令完成的。 速度命令为中间组carbon_mid的原子提供初始速度,确保这些原子的初始温度为 300 K,没有整体平移动量,妈妈是,也没有旋转动量,腐烂是。 固定 nve 应用于所有原子,以便在每一步重新计算所有原子位置,并且仅将 Berendsen 恒温器应用于 carbon_mid 组的原子 [22]。fix_modify myber 确保 fix Berendsen 使用组 carbon_mid 的温度作为输入,而不是整个系统的温度。这是确保冻结边缘不会使温度产生偏差所必需的。请注意,边的原子不需要恒温器,因为它们的运动会受到限制,见下文。 To restrain the motion of the atoms at the edges, let us add the following commands to input.lammps:
fix mysf1 carbon_top setforce 0 0 0
fix mysf2 carbon_bot setforce 0 0 0
velocity carbon_top set 0 0 0
velocity carbon_bot set 0 0 0
两个 setforce 命令分别取消施加在两条边的原子上的力。由于使用了 0 0 0,力的抵消在每一步都完成,并且沿空间的所有 3 个方向 , , 和 。这两个速度命令分别将 carbon_top 和 carbon_bot 原子沿 、 和 的初始速度设置为 0。 由于这最后四个命令,边的原子在模拟过程中将保持不动(或者至少在没有应用其他命令的情况下它们会保持不动)。
对系统施加恒定速度
速度设置命令在运行开始时施加一组原子的速度,但在整个仿真过程中不强制执行速度。当 velocity set 与 setforce 0 0 0 结合使用时,就像这里的情况一样,原子在整个模拟过程中不会感觉到任何力。根据牛顿方程,没有力意味着没有加速度,这意味着初始速度将在整个仿真过程中保持不变,从而产生恒定速度运动。
数据处理
接下来,为了测量 CNT 承受的应变和应力,让我们提取两条边之间的距离以及施加在边上的力。
variable L equal xcm(carbon_top,z)-xcm(carbon_bot,z)
fix at1 all ave/time 10 10 100 v_L file output_cnt_length.dat
fix at2 all ave/time 10 10 100 f_mysf1[1] f_mysf2[1] &file output_edge_force.dat
我们还添加一个命令,每 1000 步将原子坐标打印到 lammpstrj 文档中。
dump mydmp all atom 1000 dump.lammpstrj
最后,让我们通过使用 fix ave/time 命令打印非冻结组的温度来检查它随时间的变化:
fix at3 all ave/time 10 10 100 c_Tmid file output_temperature_middle_group.dat
关于从变量计算或者fix命令中获取数据
请注意,每条边上的力值是从两个修复 setforce mysf1 和 mysf2 中提取的,只需使用 f_ 调用它们,就像使用 v_ 调用变量和使用c_调用计算一样。
让我们运行一个小的平衡步骤,在施加任何变形之前使系统达到所需的温度:
thermo 100
thermo_modify temp Tmid
timestep 1.0
run 5000
使用 thermo_modify 命令,我们向 LAMMPS 指定我们希望在终端中打印温度,而不是整个系统的温度(由于边缘冻结,整个系统的温度无关紧要)。 让我们通过将速度设置命令与先前定义的 fix setforce 相结合,在 CNT 上施加恒定速度变形。在 input.lammps 文档中添加以下行,紧跟在上次运行 5000 命令之后:
# 2*0.0005 A/fs = 0.001 A/fs = 100 m/s
velocity carbon_top set 0 0 0.0005
velocity carbon_bot set 0 0 -0.0005
run 10000
选择的变形速度为 。的 CNT 长度随时间线性增加,正如施加的恒定速度所预期的那样。


