scCustomize:改进Seurat可视化之QC图的制作——单细胞RNA测序分析第一步
scCustomize包由来自波士顿儿童医院/哈佛医学院的博士后Samuel E. Marsh编写。该包基于Seurat,提供了若干便捷、高效的可视化方法。在所有单细胞RNA测序分析中的首要步骤之一是执行一系列QC检查和绘图,以便可以适当地筛选数据。scCustomize包含了一些可以用来快速轻松生成一些最相关QC绘图的函数。在本教程中,我们将为您演示利用SeuratData包中的HCA骨髓细胞数据完成QC绘图的全流程!本教程提供的镜像内已安装了scCustomize、SeuratData、Seurat等关键R包,快来一键运行吧!
📖 上手指南
共享协议:本作品采用知识共享署名-非商业性使用-相同方式共享 4.0 国际许可协议进行许可。
快速开始:本文档可在 Bohrium Notebook 上直接运行。点击上方的 开始连接 按钮,选择 ubuntu22-py310-r43-gpu-plot:0830镜像 和任意节点即可开始。
Warning message: “Your system is mis-configured: ‘/etc/localtime’ is not a symlink” Warning message: “It is strongly recommended to set envionment variable TZ to ‘localtime’ (or equivalent)” ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ── ✔ dplyr 1.1.2 ✔ readr 2.1.4 ✔ forcats 1.0.0 ✔ stringr 1.5.0 ✔ ggplot2 3.4.2 ✔ tibble 3.2.1 ✔ lubridate 1.9.2 ✔ tidyr 1.3.0 ✔ purrr 1.0.2 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ── ✖ dplyr::filter() masks stats::filter() ✖ dplyr::lag() masks stats::lag() ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors The legacy packages maptools, rgdal, and rgeos, underpinning the sp package, which was just loaded, will retire in October 2023. Please refer to R-spatial evolution reports for details, especially https://r-spatial.org/r/2023/05/15/evolution4.html. It may be desirable to make the sf package available; package maintainers should consider adding sf to Suggests:. The sp package is now running under evolution status 2 (status 2 uses the sf package in place of rgdal) Attaching SeuratObject scCustomize v1.1.3 If you find the scCustomize useful please cite. See 'samuel-marsh.github.io/scCustomize/articles/FAQ.html' for citation info. qs 0.25.5
Error in loadNamespace(x): there is no package called ‘hcabm40k.SeuratData’ Traceback: 1. loadNamespace(x) 2. withRestarts(stop(cond), retry_loadNamespace = function() NULL) 3. withOneRestart(expr, restarts[[1L]]) 4. doWithOneRestart(return(expr), restart)
Installing package into ‘/usr/local/lib/R/site-library’ (as ‘lib’ is unspecified)
devtools::install_github('satijalab/seurat-data')
添加线粒体和核糖体基因百分比
scCustomize包含一个简单的包装函数,可以自动将线粒体和核糖体计数百分比添加到meta.data槽中。如果您正在使用鼠标、人类、大鼠、斑马鱼、果蝇、狨猴或猕猴的数据,您只需要指定物种参数即可。
然而,对于具有不同注释的非人类/鼠标/狨猴物种,可以使用自定义前缀。只需指定物种为other,并提供您感兴趣的物种的特征列表或正则表达式模式。
注意:如果需要,请在GitHub上提交问题以获取额外的默认物种。请包括线粒体和核糖体基因的正则表达式模式或基因列表,开发者将在函数中添加额外的内置默认值。
Ensembl ID的使用
scCustomize包含所有默认物种的线粒体和核糖体基因对应的内置Ensembl ID列表。如果您的对象使用Ensembl ID作为特征名称,那么只需添加ensembl_ids参数即可。
添加细胞复杂性/新颖性QC指标
scCustomize包含一个简单的快捷函数,用于添加衡量细胞复杂性/新颖性的指标,有时可用于过滤低质量细胞。该指标通过计算log10(nFeature) / log10(nCount)的结果来计算。
注意:对于LIGER对象也有类似的函数(请参阅:Add_Cell_Complexity_LIGER),但是QC绘图函数目前仅支持Seurat对象。
绘制QC指标
scCustomize提供了许多便于使用的快速QC绘图选项。 注意:大多数scCustomize绘图函数包含...参数,允许用户提供底层使用的原始Seurat函数的任何参数。
基于VlnPlot的QC绘图
scCustomize包含4个包装了Seurat::VlnPlot()的函数。
QC_Plots_Genes() 绘制每个细胞/细胞核的基因。
QC_Plots_UMIs() 绘制每个细胞/细胞核的UMI数。
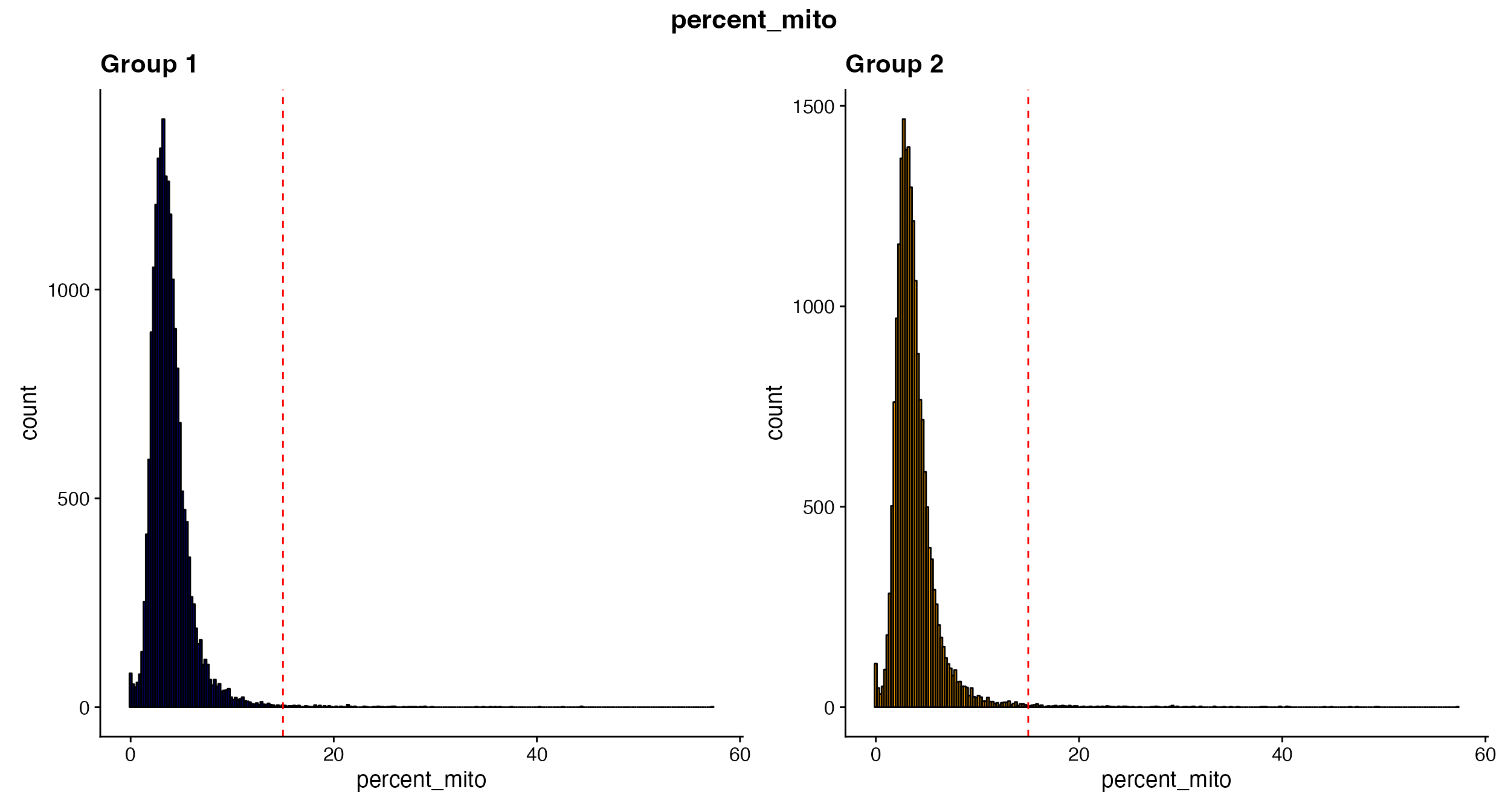
QC_Plots_Mito() 绘制每个细胞/细胞核的线粒体百分比(命名为“percent_mito”)。
QC_Plots_Complexity() 绘制每个细胞/细胞核的细胞复杂性指标(log10GenesPerUMI)。
QC_Plots_Feature() 绘制每个细胞/细胞核的“特征”。使用参数feature可以绘制对象@meta.data槽中的任何适用的命名特征。
QC_Plots_Combined_Vln() 返回QC_Plots_Genes()、QC_Plots_UMIs()和QC_Plots_Mito()的拼接图。
scCustomize函数的附加好处包括:
- 默认情况下设置要绘制的特征(除了QC_Plots_Feature)。
- 添加了高/低截止参数,以便轻松可视化潜在的截止阈值。

额外参数
除了可以使用...提供Seurat参数外,这些绘图,就像scCustomize中的许多其他绘图一样,还包含其他附加参数,用于自定义绘图输出,无需进行后续的ggplot2修改:
plot_title:更改绘图标题
x_axis_label/y_axis_label:更改轴标签。
x_lab_rotate:是否应将x轴标签旋转45度?
y_axis_log:y轴线性或log10刻度。
plot_median和median_size:在每个x轴标识处绘制中值线。
设置y_axis_log对于初始绘图非常有帮助,因为离群值会使大多数数据的可视化受到影响,而不会通过设置y轴限制来排除数据。

通过设置plot_median = TRUE参数来绘制中位数值:

组合绘图功能
作为一种快捷方式,您可以使用单个函数QC_Plots_Combined_Vln()返回三个主要QC绘图(基因、UMIs、%Mito)的单个拼贴图。

基于特征散点图的QC绘图
scCustomize包含三个函数,这些函数包装了Seurat::FeatureScatter(),并添加了潜在截止阈值的可视化以及一些其他功能:
QC_Plot_UMIvsGene() 绘制每个细胞/细胞核的基因与UMIs。
QC_Plot_GenevsFeature() 绘制基因与“特征”的关系,允许使用参数feature1绘制对象@meta.data槽中的任何适用的命名特征。
QC_Plot_UMIvsFeature() 绘制UMIs与“特征”的关系,允许使用参数feature1绘制对象@meta.data槽中的任何适用的命名特征。
新的/修改的功能:
- 更好的默认颜色调色板
- 默认为shuffle = TRUE,以防止数据集隐藏
- 能够设置和可视化潜在的截止阈值(类似于基于VlnPlot的QC绘图上面)


按连续的元数据变量着色数据
QC_Plot_UMIvsGene具有按连续的元数据变量对点进行着色的能力。这可以用于绘制%mito读数以及UMI与基因的比较。


组合绘图
如果您希望查看QC_Plot_UMIvsGene通过离散分组变量和连续变量进行绘制,而无需两次编写函数,可以使用combination = TRUE,并且绘图输出将包含两个绘图。

分析每个样本/库的中位数QC值
scCustomize还包含一些有助于返回和绘制每个样本/库上的这些指标中位数值的有用函数。
计算中位数值并返回data.frame
scCustomize包含Median_Stats函数,用于快速计算基本QC统计数据(基因、UMIs、%Mito/Cell等)的中位数并返回一个data.frame。
中位数统计函数默认存储了一些列名,但还可以使用可选的median_var参数计算其他meta.data列的中位数:
绘制中位数值
scCustomize还包含一些用于绘制这些中位数值计算的函数,可以单独使用,无需首先返回data.frame。
- Plot_Median_Genes()
- Plot_Median_UMIs()
- Plot_Median_Mito()
- Plot_Median_Other()




绘制每个样本/核糖体的细胞/核数
scCustomize还包含用于绘制每个样本中细胞或核糖体数量的绘图函数。
由于HCA骨髓数据集每个样本具有完全相同数量的细胞,因此我们将使用Analysis Plots文稿中的microglia对象。